Idée reçue : La bioinformatique, c’est l’analyse du génome humain
Le séquençage du génome humain est effectif. Seuls 10% de sa séquence restent inconnus, car inaccessibles aux techniques actuelles de séquençage, c’est-à-dire d’obtention de la succession des nucléotides le long d’une molécule d’ADN. Le résultat de ce processus physico-chimique, largement automatisé, est une longue séquence des lettres – A, C, G ou T –, initiales des motifs chimiques qui distinguent les quatre types de nucléotides. La séquence du génome humain en comporte près de 3 milliards. Seules des méthodes informatiques appropriées sont susceptibles d’analyser ce texte, d’identifier des passages pertinents (en particulier les gènes) et de les interpréter. Pour autant, la bioinformatique, ce domaine d’activité scientifique à l’interface de la biologie et de l’informatique, ne se limite pas à l’analyse des séquences génomiques humaines, loin s’en faut !
Analyser les génomes humains… et tous les autres

La double hélice, « cliché » de la bioinformatique.
UNC-CH Center for Bioinformatics
Depuis 1995, date de l’obtention de la première séquence complète du génome d’un organisme vivant, la bactérie H. influenzae, de très nombreux génomes ont été intégralement séquencés, toujours plus vite et moins cher, grâce aux progrès constants des techniques de séquençage. Autant de chaînes de caractères dont les informaticiens sont friands et pour lesquelles ils ont élaboré de puissants algorithmes et développé des programmes efficaces. Les bioinformaticiens se sont largement appuyés sur ce vaste patrimoine pour élaborer des méthodes d’analyse des « textes » génomiques. Il s’agit d’identifier les régions codantes des gènes, qui portent l’information utilisée par la cellule pour synthétiser les protéines, mais aussi les régions impliquées dans la régulation de l’expression de ces gènes. Ou de rechercher des sous-séquences qui se répètent au sein du génome, ou encore des sous-séquences qui forment des palindromes, etc.
Analyser, mais pas seulement les génomes
Produits des gènes, les molécules d’ARN, chaînes de ribonucléotides, et les protéines, chaînes d’acides aminés, acquièrent leurs fonctions en se repliant dans l’espace. La détermination expérimentale de cette structure tridimensionnelle reste un processus long et coûteux. Sa prédiction à partir de la séquence constitue de ce fait un défi pour la bioinformatique, et ce depuis plusieurs dizaines d’années, alors même que se trouve contestée l’hypothèse que toute l’information déterminant ce repliement se trouverait dans la séquence…
Outre ce problème critique, la bioinformatique dite structurale a développé de nombreux algorithmes et programmes destinés à analyser, visualiser et manipuler ces structures moléculaires tridimensionnelles.
Au-delà de l’analyse : structurer et diffuser les connaissances
Les séquences, ainsi que les informations qu’elles portent ou auxquelles elles sont associées, sont répertoriées, quand elles sont rendues publiques, dans de grandes bases de données. Ainsi, face à une nouvelle séquence d’intérêt, la première action d’un biologiste est désormais de la comparer au contenu de ces bases pour retrouver d’éventuelles séquences similaires, auxquelles peuvent être attachées des informations susceptibles de le renseigner sur la nature et les fonctions de sa séquence. Cette recherche de séquences similaires repose sur l’emploi de programmes de la famille Blast.
Les bases de données sont progressivement devenues le moyen privilégié pour la communauté des chercheurs en biologie de rassembler, structurer, croiser et diffuser les données et les connaissances qu’ils acquièrent sur les organismes vivants. La consultation du numéro spécial annuel de la revue NAR (Nucleic Acids Research) consacré aux bases de données permet de mesurer leur diversité et leur intérêt stratégique.
Explorer de nouveaux champs de recherche
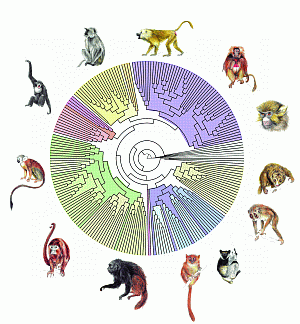
Arbre phylogénétique incorporant 217 des 233 espèces actuelles de primates, reconstruit par l’algorithme PhyML, conçu par Stéphane Guindon et Olivier Gascuel.
© E. Douzery, P.-H. Fabre (CNRS / ISEM), O. Gascuel, S. Guindon (CNRS / LIRMM) et F. Chevenet (IRD)
Les progrès des techniques de séquençage permettent également des démarches systématiques inimaginables il y a encore quelques années. La métagénomique consiste ainsi à séquencer tout l’ADN présent dans un échantillon, que ce soit une goutte d’océan ou un prélèvement du contenu de l’estomac. L’objectif est alors d’identifier les micro-organismes présents grâce à ces fragments de leur ADN. Par ailleurs, la disponibilité de la séquence de nombreuses souches d’un même organisme microbien permet de déterminer ce qui les différencie, en vue, par exemple, de caractériser leur aptitude à causer une maladie.
La disponibilité de séquences relatives à un nombre sans cesse croissant d’organismes différents modifie profondément la problématique de la reconstruction des arbres phylogénétiques. Ces arbres – au sens informatique du terme – rendent compte de l’histoire évolutive de ces espèces. Les algorithmes les plus récents sont capables de comparer plusieurs dizaines de milliers de séquences d’un même gène chez autant d’organismes et de proposer un arbre qui rende compte au mieux des similarités et dissimilarités, et de ce fait de la proximité évolutive des espèces.
Étudier la dynamique des interactions
Les molécules ne peuvent être analysées de façon isolée. L’expression d’un gène, c’est-à-dire l’exploitation par la machinerie cellulaire de l’information qu’il porte, varie dans le temps, régulée par le produit d’autres gènes, eux-mêmes sous l’influence d’autres produits de gènes. Les « puces à ADN » et autres dispositifs expérimentaux permettent de mesurer ces niveaux d’expression, en fonction de conditions environnementales contrôlées. Les données qu’elles produisent font l’objet d’analyses statistiques.
Les interactions moléculaires composent donc des réseaux complexes que les biologistes cherchent à reconstituer afin d’en comprendre la dynamique. Ces réseaux peuvent être représentés par des graphes, troisième classe d’objets, après les chaînes de caractères et les arbres, que les informaticiens connaissent bien. Mais l’étude de la dynamique des interactions nécessite le recours à des modèles appropriés, des plus simples – réseaux booléens –, aux plus complexes – systèmes d’équations différentielles, voire aux dérivées partielles, ou encore simulation stochastique.
Sans prétendre à l’exhaustivité, ce panorama rapide de la bioinformatique laisse entrevoir un domaine de recherche motivé par des problématiques biologiques extrêmement diverses et empruntant à de nombreux domaines de l’informatique fondamentale et des mathématiques appliquées : algorithmique, modélisation des données et des connaissances, probabilités et statistiques, optimisation combinatoire…
L’analyse des génomes humains, si elle est porteuse d’espoirs thérapeutiques et d’avancées scientifiques enthousiasmantes, ne constitue vraiment qu’une partie de l’activité des bioinformaticiens.
Newsletter
Le responsable de ce traitement est Inria. En saisissant votre adresse mail, vous consentez à recevoir chaque mois une sélection d'articles et à ce que vos données soient collectées et stockées comme décrit dans notre politique de confidentialité
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !
