Les séquenceurs à haut débit

Photo © CNRS Photothèque – Hubert Raguet.
Le génome d’un être vivant est une suite de petites molécules, les nucléotides, de quatre types différents, notés par les initiales A, C, G et T de leurs bases azotées. La succession de ces nucléotides constitue la molécule d’ADN. Au sein de chacune des cellules de tout organisme vivant, l’ADN code ainsi l’information nécessaire à l’organisme pour se développer, se maintenir et se reproduire. En temps normal, toutes les cellules d’un même individu ont exactement, ou presque, le même génome. De plus, ce génome est identique à plus de 99% au sein de l’espèce humaine. Les génomes font de quelques milliers de bases pour les virus et quelques millions pour les bactéries (4,6 mégabases (Mb) pour la bactérie Escherichia coli) à quelques milliards pour les organismes pluri-cellulaires (3,4 gigabases (Gb) pour l’homme).
Le séquençage est la lecture de la succession des nucléotides le long d’une molécule d’ADN. Le résultat produit est un texte écrit dans l’alphabet A, C, G, T. Les séquenceurs lisent des fragments de plusieurs bases, appelés reads, dont la longueur varie en fonction des technologies. Le génome s’obtient ensuite par assemblage des reads.

Séquençage de génome par assemblage de reads.
L’assemblage des trois reads, par la mise en correspondance de leurs points communs, permet d’obtenir une séquence plus longue. C’est le point de départ de l’obtention de la séquence d’un génome complet.
Depuis 2005, les nouvelles technologies de séquençage ont révolutionné ce domaine, et soulèvent de nouveaux défis aux informaticiens. Nous présentons ici les techniques de séquençage, leurs utilisations, et quelques problèmes et solutions informatiques s’y rapportant.
Le séquençage d’ADN
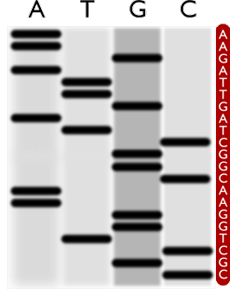
Séquençage par méthode de Sanger, produisant ici un read de 21 bases.
Méthode de Sanger
La méthode de séquençage de Sanger a été mise au point en 1976. La séquence d’ADN est polymérisée en quatre ensembles de fragments de différentes tailles, chaque ensemble terminant sur une des 4 bases. Les fragments migrent ensuite dans un gel, ce qui permet de détecter leur longueur et de lire la séquence. Cette méthode a été optimisée et automatisée durant plus de 25 ans. C’est ainsi qu’en 1995, le premier génome d’un organisme vivant, la bactérie H. influenzae, a été entièrement séquencé et qu’en 2001, une première version du génome de référence pour l’espèce humaine était obtenue. Un séquenceur mettait alors près de 10 heures pour produire des reads de 500 à 600 bases dans 96 tubes de verre de quelques microns de diamètre appelés capillaires. Son débit était donc d’environ 115 kilobases (Kb) par jour.
Les séquenceurs à haut débit
Au début des années 2000, quelques études proposent de nouvelles technologies qui révolutionnent le séquençage. Il ne s’agit plus de mesurer la longueur de certains fragments, mais de lire directement la séquence d’ADN, base par base, par cycles successifs. Ces recherches ont été extrêmement vite transférées, et des séquenceurs à haut débit ont été proposés sur le marché à partir de 2004 (voir tableau ci-dessous). Toutes ces techniques traitent les bases une par une, en alternant des réactions chimiques couplées à une détection optique et des phases de nettoyage.
| Société | Roche | Applied Biosystems | Illumina |
| Séquenceur | 454 Titanium | Solid 5500 | HiSeq 2000 |
| Technique | Incorporation de bases et lumière produite par oxydation de la luciférine. | Ligation de sondes contenant des paires de bases. | Incorporation de bases étiquetées par des couleurs différentes. |
| Longueur des reads | ~ 400 bases | ~ 75 bases | ~ 100 bases |
| Erreurs fréquentes | insertions | substitutions | substitutions |
| Débit annoncé | ~ 1 Gb / jour | ~ 10 Gb / jour | ~ 50 Gb / jour |
Comparaison des caractéristiques de différents séquenceurs à haut débit.
Évolution du coût et de la capacité
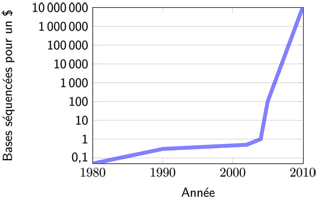
Le coût du séquençage a été considérablement réduit ces dix dernières années.
Par ces nouvelles techniques, les séquenceurs à haut débit ont permis une vraie rupture dans les quantités de données produites, qui font plus que décupler chaque année ! Ces séquenceurs coûtent aujourd’hui plusieurs centaines de milliers d’euros à l’achat, auxquels il faut ajouter le coût des réactifs. Une expérience (un run) nécessite quelques milliers d’euros de matériel. La préparation des échantillons prend plusieurs heures voire plusieurs journées.
Le plus grand centre de séquençage au monde, le Beijing Genome Institute (BGI), en Chine, possède actuellement 137 Illumina HiSeq 2000 et 27 AB Solid 4.0. En France, le Genoscope, à Évry, est le principal centre de séquençage, mais d’autres plateformes, à Paris et en région, ont des capacités intéressantes.
De nouvelles technologies sont attendues dans les prochaines années, pour améliorer le débit et la qualité des résultats, et baisser les coûts et le temps de préparation. Par exemple, la société Ion Torrent, rachetée par Roche en 2010, propose une technique de séquençage couplée à un microprocesseur sur silicium, qui permet de se passer de l’étape de détection optique.
Séquencer, pour quoi faire ?
Utilisation directe
Le séquençage du premier génome humain, de 1998 à 2003, n’a pas arrêté les besoins de séquençage, bien au contraire. Aujourd’hui, de nombreuses études se focalisent sur le polymorphisme, c’est-à-dire sur les variations entre plusieurs individus. Le projet 1000 Genomes, débuté en 2008, a pour objectif de séquencer le génome de 2500 personnes, afin d’identifier les spécificités de chacun. Avant la fin de notre décennie, nous arriverons au génome à 1000 $, et ce coût peut encore baisser. Cela rend possible la médecine personnalisée, le but étant de prescrire des traitements adaptés aux spécificités génétiques de chacun et à leurs impacts, par exemple sur le métabolisme. Dans ces applications, on parle de reséquençage, car la séquence de chaque individu est très proche du génome de référence de l’espèce.
En dehors de l’homme, seulement un millier d’espèces a été séquencé, ce qui est peu comparé aux millions d’espèces connues. De plus, une nouvelle discipline a fait son apparition dans les années 2000 : la métagénomique étudie des séquences d’ADN d’un écosystème dans son ensemble, sans séparation des espèces ni des individus, que cela soit dans des milieux naturels (océan, terre) ou bien chez l’homme (flore intestinale). Étudier des métagénomes permet aussi de découvrir de nouvelles séquences, parfois provenant d’espèces inconnues.
Utilisations dérivées
Les résultats du séquençage ouvrent d’autres perspectives de recherche. Lorsqu’on connaît déjà un génome de référence, par exemple le génome humain, le séquençage sert à obtenir un ensemble de reads qui, une fois localisés, traduisent un phénomène biologique. On passe ainsi du séquençage au décryptage du génome.
Par exemple, les études de transcriptomique examinent tous les ARN produits par une cellule à un instant donné. En fonction du nombre de reads localisés à un même endroit, il est possible de comprendre quelles sont les parties du génome les plus actives en fonction des différents tissus (comme le rein, le cerveau ou la rétine) et de leur environnement.
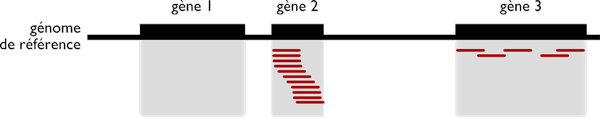
Les techniques de transcriptomique par RNAseq permettent de mesurer les niveaux d’expression de différents gènes, en localisant les reads issus d’ARN messager sur le génome. Ici, le gène 2 est fortement exprimé, tandis que le gène 3 l’est faiblement.
D’autres études concernent les sites de fixation des facteurs de transcription, ou bien la chromatine (la structure à grande échelle de l’ADN). Dans de nombreuses applications, le séquençage à haut débit est ainsi en train de remplacer d’autres techniques moins flexibles telles que les puces à ADN.
Défis algorithmiques
Algorithmique du texte
La bioinformatique des séquences est un bel exemple de transfert de recherche fondamentale en combinatoire vers une recherche appliquée. En effet, la recherche de mots dans un texte est un problème déjà ancien d’algorithmique du texte, étudié dès les années 1970. Aujourd’hui, ces techniques sont utilisées dans les traitements de texte (pour rechercher un mot dans une page) ou dans les moteurs de recherche (pour rechercher les pages web où apparaît un mot). Un read n’étant qu’un fragment d’un génome, il est généralement possible de retrouver sa localisation d’origine si on dispose du génome de référence de l’espèce. Dans toutes ces situations, il s’agit donc de trouver des occurrences d’une courte chaîne dans un très long texte, le texte pouvant être un ensemble de pages web ou bien un ou plusieurs génomes.
Mutations
Il est souvent utile de faire des recherches approximatives, c’est-à-dire de retrouver la courte chaîne recherchée, y compris avec des erreurs, comme par exemple des erreurs orthographiques. Pour les séquences d’ADN, les différences entre chaînes similaires sont appelées les mutations.
Lors de chaque division de cellule, ces mutations arrivent à une fréquence d’environ 10−5 par base, mais n’ont généralement aucune conséquence sur la santé de l’individu. Chacun d’entre nous ne possède en général que quelques dizaines de mutations par rapport au patrimoine génétique légué par ses deux parents. Dans la population humaine, deux humains ont environ 0,5% de différences dans leur génome. À plus grande échelle, ces mécanismes de mutations sont le support de l’évolution, et se retrouvent dans les séquences : nous avons plus de 95% de notre génome en commun avec les grands singes, mais beaucoup moins avec les bactéries. Un read, puisqu’il provient du génome d’un individu, peut donc avoir des différences avec le génome de référence de l’espèce.
Ces différences concernent le plus souvent la substitution, l’insertion ou la suppression d’une seule base, mais il peut aussi y avoir des fragments plus longs insérés ou supprimés. De plus, les reads peuvent contenir des erreurs de séquençage en raison de la façon dont ils sont produits : les séquenceurs ne sont pas parfaits.

Recherche approximative de texte et localisation de reads dans un génome.
Plus un read contient de mutations par rapport au génome de référence, plus il est difficile à localiser. Mais, paradoxalement, plus il est potentiellement important pour les biologistes ou les médecins. Les mutations sont très importantes afin de comprendre l’origine génétique de certaines maladies. Cela est d’autant plus vrai pour les cellules cancéreuses, qui connaissent souvent des phénomènes d’hyper-mutations, entraînant de nombreuses différences avec le génome de référence, y compris la création de chimères par échanges génétiques entre deux parties du génome.
Solutions algorithmiques
Les problèmes bioinformatiques tels que la localisation des reads sur un génome de référence peuvent se résoudre par différentes techniques algorithmiques. Pour rechercher rapidement des informations, une solution consiste à indexer le texte initial. Ainsi, certains livres contiennent un index. Son rôle est d’indiquer, pour certains mots-clés choisis, les endroits auxquels ils se trouvent dans l’ouvrage.
S’il est difficile de définir ce qu’est un mot-clé pour une séquence d’ADN (qui n’est représentée que par une suite de A, C, G et T), des approches similaires sont néanmoins utilisées.
Indexation des k-mots
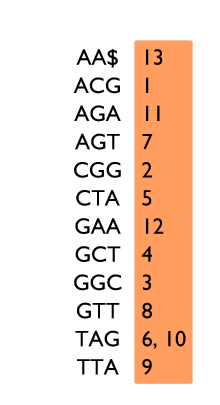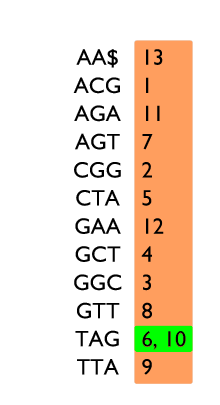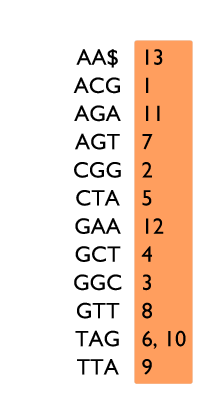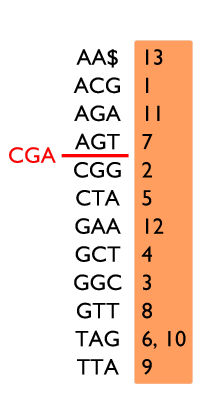
Table de 3-mots sur le texte ACGGCTAGTTAGAA$, avec deux exemples de recherche.
Pour les séquences d’ADN, on peut par exemple indexer l’ensemble des suites de k lettres, suites appelées k-mots. Pour rechercher une chaîne quelconque, on extrait tous les k-mots de la chaîne à rechercher et on utilise l’index pour connaître la position de tous ces k-mots dans le texte. Ensuite, la comparaison est effectuée dans le voisinage de chacun des k-mots, afin de vérifier que la totalité de la chaîne recherchée est bien présente dans le texte, éventuellement avec un certain nombre de mutations. Une telle approche est par exemple utilisée par BLAST (inventé en 1990), qui est certainement l’application bioinformatique la plus utilisée à ce jour. L’article proposant BLAST est d’ailleurs l’un des articles scientifiques les plus cités de tous les temps : des dizaines de milliers de scientifiques ont utilisé BLAST dans leurs travaux.
Un exemple d’indexation par k-mots est donné sur la figure à gauche. Pour chaque 3-mot du texte ACGGCTAGTTAGAA$, nous enregistrons à quelle position il apparaît. Par exemple le 3-mot GAA apparaît en position 12 dans le texte. Les 3-mots sont rangés dans l’ordre alphabétique afin de permettre une recherche plus rapide, comme dans un dictionnaire.
Pour chercher un mot dans un dictionnaire, sans aucune information sémantique, ne première méthode est de tourner les pages une à une. S’il y a 1 000 pages, il faut entre 1 et 1 000 gestes suivant que le mot commence par A ou Z. Cela est fastidieux, mais de cette façon on est sûr de ne pas manquer le mot recherché.
Les informaticiens savent faire mieux : ouvrir le dictionnaire en son milieu ; si le mot est situé avant le milieu, ne considérer que la première moitié des pages, s’il est situé après, ne considérer que la deuxième moitié ; ouvrir le dictionnaire au milieu de la moitié des pages considérées ; répéter le procédé jusqu’à ce qu’il ne reste qu’une page. Que se passe-t-il ? À chaque fois, on ne considère que la moitié des pages, donc 1 000, 500, 250, 125, 63, 32, 16, 8, 4, 2, 1 page… et hop le mot est trouvé ! en 10 étapes au lieu de 1 000 ! Voilà un bel algorithme, « optimal », on ne peut pas être plus efficace, et qui « converge », diviser sans cesse le nombre de pages ne peut que finir par donner 1. Presque tous les algorithmes de recherche sont construits sur de tels mécanismes.
Quand un k-mot est recherché, TAG par exemple, il faut d’abord vérifier s’il est présent dans l’index. Dans notre exemple, il est trouvé. L’information stockée dans l’index permet directement de savoir qu’il apparaît aux positions 6 et 10.
En imaginant que ce 3-mot TAG appartienne à un read plus grand (GTTAG), trouver TAG aux positions 6 et 10 permet de déduire que GTTAG peut apparaître en positions 4 et 8. Une comparaison lettre à lettre permet d’observer que GTTAG apparaît avec une substitution en position 4 et qu’il apparaît exactement en position 8.

En revanche, la recherche de CGA échoue : il devrait se trouver entre AGT et CGG dans l’index, mais il n’y est pas. Cela signifie donc que CGA n’apparaît pas dans le texte.
Arbre des suffixes
L’arbre des suffixes, proposé par Weiner en 1973, est une autre solution pour indexer des données. Il s’agit d’une structure dans laquelle tous les suffixes du texte sont enregistrés, c’est-à-dire toutes les chaînes du texte commençant à n’importe quel endroit du texte et se terminant à la fin. Le mot Bonjour a ainsi 8 suffixes : Bonjour, onjour, njour, jour, our, ur, r et le mot vide.
Un arbre des suffixes permet de rechercher n’importe quelle sous-chaîne du texte en un temps proportionnel à la longueur de cette sous-chaîne. Ce temps est donc indépendant de la longueur du texte indexé. Autrement dit, en théorie, la recherche d’une sous-chaîne prendrait un temps identique, qu’on la recherche dans un texte d’une dizaine de pages ou dans l’ensemble des textes de la planète.
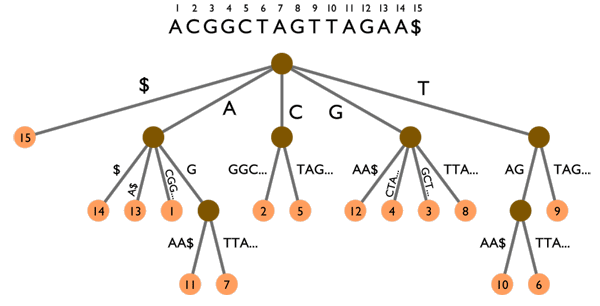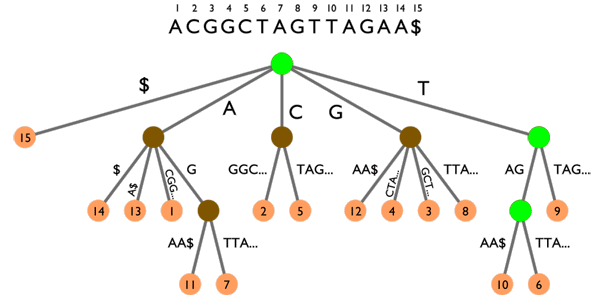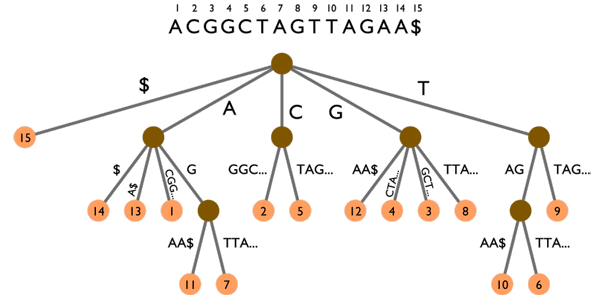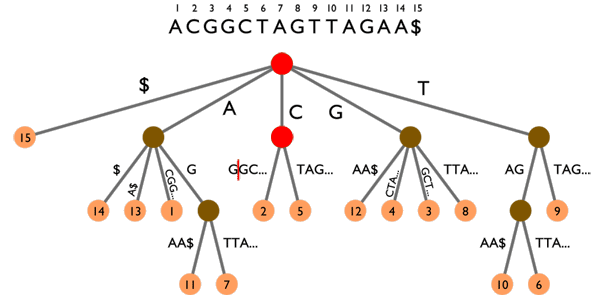
Arbre des suffixes sur le texte ACGGCTAGTTAGAA$. Les animations montrent deux exemples de recherche :
TAG (points verts) et CGA (points rouges). Voir les explications dans le texte ci-dessous.
L’arbre des suffixes peut aussi être utilisé pour rechercher des k-mots, comme l’illustre la figure ci-dessus. Suivre un chemin de la racine de l’arbre (le point marron en haut) jusqu’à une « feuille » (l’un des points orange en bas) permet de lire un suffixe du texte. Par exemple, en partant de la racine et en descendant jusqu’à la feuille 12, on peut lire le suffixe GAA$ qui est effectivement celui commençant en position 12 dans le texte.
La recherche de mots se fait toujours en partant de la racine. Rechercher TAG (points verts) nous amène dans un « nœud » duquel sortent deux « branches » amenant aux feuilles 10 et 6 (points jaunes). TAG apparaît donc deux fois dans le texte, en positions 10 et 6. D’un autre côté, rechercher CGA (points rouges) fonctionne pour les deux premières lettres (CG) mais échoue sur la dernière lettre : seul CGG existe dans le texte.
L’arbre des suffixes est un index beaucoup plus puissant que la table des k-mots et il permet d’effectuer des opérations beaucoup plus complexes. Ainsi, l’arbre des suffixes permet de connaître la plus longue chaîne commune à deux textes, ou encore de détecter des répétitions dans un texte. Ce type de calcul est utilisé en bioinformatique afin de savoir quelles sont les parties qui ont été les plus conservées entre deux génomes d’espèces différentes, et a aussi des applications dans d’autres domaines comme la détection de plagiat.
Nouvelles méthodes
De nombreuses recherches ont amélioré les index de k-mots et les arbres de suffixes, en proposant des structures plus pertinentes pour certains problèmes. Les méthodes que nous développons actuellement ne se contentent plus de rechercher un mot en autorisant quelques erreurs, mais permettent aussi de trouver des informations qui sont éventuellement distantes sur le génome de référence, afin de découvrir des échanges génétiques. Au lieu de considérer le read comme un seul mot devant être trouvé de manière contiguë sur le génome, on peut alors le considérer comme un ensemble de segments plus courts pouvant se trouver à différents endroits du génome, segments pouvant eux-mêmes contenir des mutations.
Évolutions technologiques
Même les meilleurs algorithmes nécessitent de stocker et de traiter de grandes quantités de données. Le problème actuel de la bioinformatique est que l’explosion des données (dont la quantité est multipliée par 10 tous les ans) se fait à un rythme bien supérieur à l’évolution technologique, qui suit la loi de Moore (qui prévoit un doublement du nombre de transistors tous les 18 mois).
Un point important concerne l’adéquation entre les modèles théoriques de complexité de calcul et les implémentations. Par exemple, les différents niveaux de mémoire jouent un rôle important. Accéder au cache de premier niveau d’un processeur est 100 à 1000 fois plus rapide qu’accéder à la mémoire centrale d’un ordinateur, et 104 à 106 fois plus rapide que d’accéder à un disque dur.
Cet élément est d’autant plus à prendre en compte que certaines structures d’indexation ont besoin de beaucoup de mémoire : un arbre des suffixes nécessite au moins 10 octets par caractère du texte indexé. L’indexation d’un génome humain, qui est composé d’environ trois milliards de lettres, nécessite des dizaines de Go avec l’arbre des suffixes. Il existe néanmoins d’autres structures, compressées, qui apportent des compromis entre efficacité et place mémoire.
Enfin, des projets actuels de séquençage tablent sur l’acquisition d’environ 1 pétaoctet de données, soit un million de milliards de caractères. Comment parvenir à analyser ces données massives, dont on a du mal à se figurer le gigantesque volume ? Les solutions apportées sont à la fois algorithmiques et technologiques, par exemple en utilisant du parallélisme. Cependant, les algorithmes qui fonctionnent bien en séquentiel, sur un seul processeur à la fois, ne sont pas forcément ceux qui se parallélisent le mieux. Une voie importante de recherche est ainsi l’adaptation des algorithmes existants et le développement de nouveaux pour tirer parti des nouvelles architectures et de ces nouvelles données.
Une première version de cet article est parue dans le numéro 143 du mensuel PROgrammez.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

