

sous licence Creative Commons
Éclairer les origines de la COVID-19 à partir de l’analyse comparée des génomes viraux
Or, enquêter sur l’origine de la COVID-19 repose en premier lieu sur une analyse minutieuse des génomes de souches virales collectées lors des premiers stades de ce qui n’était alors qu’une épidémie. Cette enquête met en jeu des outils bien connus dans le domaine de la bio-informatique avec, aux avant-postes, l’analyse phylogénétique. Cette dernière vise à retracer l’histoire évolutive de lignées virales en reconstituant, à partir de la modélisation mathématique, les évènements évolutifs qui ont abouti aux génomes des virus analysés. Nous verrons par la suite comment ce type d’analyse permet de répondre clairement à certaines questions liées aux origines du SARS-CoV-2.
PRRAR : cinq lettres déterminantes
Ces cinq lettres, PRRAR, cristallisent bon nombre des interrogations autour des origines du SARS-CoV-2. Elles correspondent à la suite d’acides aminés — les briques qui constituent les protéines — d’une région bien particulière de la protéine de spicule du SARS-CoV-2. Cette spicule est véritablement la clef utilisée par le virus pour faire pénétrer son génome dans la cellule humaine et lui emprunter son système de réplication de l’ADN. La protéine spicule du SARS-CoV-2 fait montre d’une redoutable efficacité qui s’explique en grande partie par la présence du motif PRRAR, correspondant au site dit de « clivage » par la furine. La furine, une protéine présente naturellement chez l’homme, va en effet « ouvrir » (les biologistes moléculaires disent « cliver ») la spicule, précisément au niveau du motif PRRAR, et ainsi l’activer. La spicule peut alors s’arrimer à la surface de nos cellules avant d’y injecter le génome viral.
La présence du motif de clivage par la furine peut être considérée par certains comme une sorte d’anomalie évolutive. En effet, ce motif est absent des séquences de spicules observées au sein des génomes les plus similaires au SARS-CoV-2 connus à l’heure actuelle. On le trouve cependant chez certains virus relativement distants d’un point de vue évolutif, circulant au sein de chauve-souris et affectant les voies respiratoires d’autres espèces de mammifères, dont l’humain. Mais ces derniers virus ne peuvent pas être les ancêtres directs du SARS-CoV-2, ou tout au moins pas exclusivement (voir les figures ci-après), car leur génome est en moyenne bien plus divergent que celui d’autres souches virales circulant, elles-aussi, chez la chauve-souris et d’autres mammifères.
Alors comment expliquer la présence de ce motif chez le SARS-CoV-2 ? Comment expliquer que certaines régions de ce génome sont très similaires à des séquences trouvées tantôt chez la chauve-souris, tantôt chez le pangolin ? Doit-on recourir à l’hypothèse d’une fuite d’un laboratoire d’une chimère virale créée par l’homme pour faire sens des observations disponibles ou est-il possible que les mécanismes classiques de l’évolution moléculaire fournissent une explication tout à fait plausible ? Comme nous allons le voir, avancer sur ces questions nécessite de reconstruire puis d’interpréter les arbres de l’évolution.
Des grottes du Yunnan à Wuhan…
Le SARS-CoV-2 a été nommé ainsi en février 2020. Mais les ancêtres de ce virus, plus ou moins semblables à celui-ci, sont étudiés depuis de nombreuses années déjà. Ainsi, le SARS-CoV (ou SRAS-CoV dans sa version française) a sévi sous forme épidémique en 2002-2003 suite à la consommation par l’homme de viande de civette infectée. Il a causé la mort de près de 800 personnes. Le MERS-CoV a été quant à lui détecté pour la première fois en 2012. À ce jour, il est responsable de près de 600 décès dans 26 pays. Sa transmission se poursuit à faible intensité. Une multitude d’autres virus de la famille des coronavirus circulent donc très probablement à l’heure actuelle au sein de plusieurs espèces animales.
Les chauve-souris constituent vraisemblablement le principal réservoir pour les coronavirus. Bien que les zoonoses, c’est-à-dire les transmissions de l’animal à l’homme, impliquent fréquemment d’autres espèces, il est techniquement possible pour les chauve-souris de transmettre des virus de type coronavirus à l’homme sans intermédiaire. La quête des origines de la COVID-19 s’est donc naturellement orientée très tôt sur les similarités entre séquences du SARS-CoV-2 et celles de coronavirus infectant des espèces susceptibles de transmettre le virus à l’homme, avec la chauve-souris comme suspect principal.
Il est ainsi apparu que le génome viral le plus proche (en moyenne) de celui du SARS-CoV-2 correspondait à une souche nommée RaTG13. Cette souche est l’une des 293 récoltées en 2013 dans des excréments de chauve-souris occupant une ancienne mine dans la province chinoise du Yunnan. Cette zone avait en effet fait l’objet d’études de la part de Shi Zhengli, biologiste diplômée de l’université de Montpellier en 2000 et directrice du « Center for Emerging Infectious Diseases » à l’Institut de Virologie de Wuhan (WIV), parfois surnommée « Bat Woman » par les journalistes. En 2012, six travailleurs chargés de nettoyer l’ancienne mine de cuivre en question étaient subitement tombés malades, présentant des symptômes de type pneumonie aiguë. Trois de ces mineurs sont décédés des suites de cette maladie. Plus de huit ans après cet épisode, les premiers cas de COVID-19 voyaient le jour à proximité du marché de Wuhan, à plus de 1800 km des grottes du Yunnan mais à seulement quelques encablures du WIV…
De fortes suspicions se sont donc portées sur le WIV et l’équipe de Shi Zhengli en particulier. La fameuse hypothèse d’un accident de manipulation dans l’enceinte de l’Institut (« lab leak »), responsable de la première transmission à l’homme du SARS-CoV-2, a suivi un itinéraire atypique au sein de la communauté scientifique et au-delà. Rapidement considérée comme une hérésie, certains chercheurs ont concédé plus tard que l’hypothèse d’une fuite de laboratoire avait été mise à l’écart peut-être trop hâtivement et de manière trop catégorique. Cette évolution dans l’attitude du monde scientifique vis-à-vis de l’hypothèse du « lab leak » s’explique, au moins en partie, par le fait que le « chaînon manquant » entre RaTG13, qui a transité par le WIV, et le SARS-CoV-2 n’a toujours pas été identifié.
Composition d’une mosaïque génomique
Mais ce chaînon existe-t-il vraiment ? Ou plutôt, est-il unique ? Nous savons en effet que certaines chauve-souris peuvent être infectées par plusieurs souches distinctes de coronavirus. Il est également possible que des évènements d’infections multiples se produisent chez d’autres animaux cibles de ces virus tels que les pangolins par exemple. En cas de co-infection, différents génomes peuvent « recombiner » et donner naissance à un nouveau génome viral mosaïque, une chimère des génomes viraux d’origines distinctes. Les recombinaisons permettent parfois de générer des génomes composites d’une efficacité accrue. Les analyses des premiers génomes du SARS-CoV-2 apparus à l’hiver 2019-2020 ont ainsi donné lieu à des hypothèses contradictoires concernant l’espèce responsable de la transmission du virus à l’homme. En effet, certaines parties du gène de la spicule du SARS-CoV-2 sont plus semblables aux séquences de coronavirus circulant chez le pangolin comparées à celles de la chauve-souris. Une analyse phylogénétique permet de comprendre cette contradiction apparente, comme nous allons le voir.
Le graphe de transmission (voir figure ci-dessous) présente un scénario possible d’évolution du virus circulant au sein de différentes espèces. On s’intéresse ici à deux régions du génome (régions verte et mauve dont les positions figurent sur le schéma du chromosome en haut à droite de la figure). La suite des évènements de transmission peut être représentée sous la forme d’un graphe où chaque ligne verticale correspond à une lignée, chacune décrivant l’histoire évolutive d’une région du génome. Les lignes horizontales, qui correspondent aux nœuds du graphe, sont des évènements de transmission du virus. Lors d’un tel événement, le génome complet du virus (incluant donc les deux lignées d’intérêt ici) est dupliqué : la copie d’origine (située au point de départ de chaque flèche horizontale) reste au sein de l’hôte responsable de l’infection tandis qu’une nouvelle copie vient occuper l’hôte nouvellement infecté (au point d’arrivée de chaque flèche). À noter qu’un des évènements de transmission se fait d’une lignée infectée vers une autre, elle aussi déjà infectée (flèche horizontale bleue, à partir du nœud 4). Ce cas de figure correspond à une co-infection et permet de redistribuer le matériel génétique via le phénomène de recombinaison de deux souches virales distinctes cohabitant au sein d’un même hôte. Ainsi, à l’issue de la recombinaison, la région verte est héritée de la lignée RaTG13 (notée C-S(RaTG13)) tandis que la région en mauve provient de l’autre chauve-souris (la chauve-souris « ancestrale », notée C-S(ancêtre)).
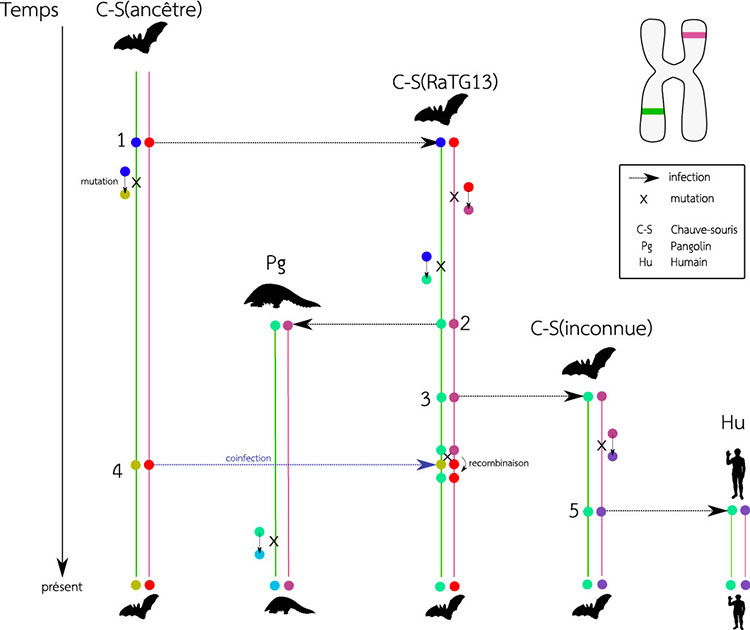
Un exemple de graphe de transmission.
Le graphe de transmission ci-dessus représente la réalité du processus d’infection de différents hôtes (trois chauve-souris, un pangolin et un humain ici) par autant de souches virales. La suite des événements de transmissions du virus et de mutations au sein de son génome constitue une source d’information très précieuse pour comprendre à la fois la dynamique de l’infection mais aussi ses origines. Que peut-on dire alors de ce graphe à partir des données issues du séquençage de génomes viraux contemporains ? Les arbres phylogénétiques (voir figure ci-après) proposent des éléments de réponse, ils décrivent l’histoire évolutive de nos deux régions du génome viral. Ces arbres sont « inscrits » dans le graphe de transmission : pour chaque région et donc chaque couleur (vert et mauve), parcourir le graphe à partir des feuilles, en remontant vers la racine donne l’arbre correspondant. Notons ici que les hôtes figurant au sein du graphe de transmission ne sont pas tous représentés au sein des arbres phylogénétiques. Ainsi, nous considérons dans notre exemple que le génome viral infectant la chauve-souris marquée d’un point d’interrogation (notée C-S(inconnue) dans le graphe de transmission) ne figure pas au sein des bases de données génomiques. Ici, seuls les génomes qui correspondent à des lignées connues (la chauve souris « ancêtre », le pangolin, la chauve-souris RaTG13 et l’homme) sont utilisés pour reconstruire les phylogénies.
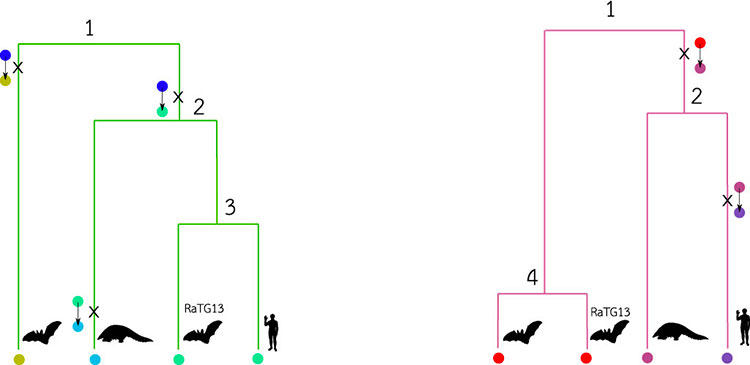
Arbres phylogénétiques.
Lorsque l’on retrace l’évolution de la région mauve en suivant « en arrière dans le temps » les quatre lignées échantillonnées, l’arbre phylogénétique montre que la souche ayant infecté le pangolin est la plus proche de celle de l’homme. Pour la région verte en revanche, c’est bien RaTG13, la souche de la grotte du Yunnan, qui est la plus proche de la lignée responsable de l’infection chez l’homme. Ainsi, certaines régions indiquent que les souches circulant chez les pangolins sont les plus proches parentes du SARS-CoV-2 circulant chez l’homme, tandis que d’autres régions suggèrent que ce sont les chauve-souris qui ont transmis le virus à l’homme. L’analyse bio-informatique de la diversité génétique au temps présent permet donc, grâce à des modèles probabilistes décrivant l’évolution à l’échelle moléculaire, de remonter dans le temps pour mieux comprendre le mode et le tempo de l’évolution virale. La reconstruction phylogénétique à l’échelle de génomes complets constitue ainsi un instrument puissant pour découvrir l’origine d’une pandémie.
…de Wuhan au Laos.
Bien que l’analyse phylogénétique et la reconstruction de graphes de transmission nous permettent de comprendre l’origine de la diversité génétique le long des quelques 30 000 nucléotides qui composent le génome du SARS-CoV-2, il n’en reste pas moins que RaTG13 a transité par le WIV, alimentant ainsi les doutes sur les origines de la COVID-19. Cependant, en septembre 2021 une équipe de l’Institut Pasteur a rendu publique la découverte de trois nouvelles souches de coronavirus plus proches de SARS-CoV-2 que ne l’est RaTG13. Ces souches, baptisées BANAL, ont été prélevées au Laos au cours de l’été 2020. Cette découverte est importante car elle montre qu’il existe des souches très proches du SARS-CoV-2 d’un point de vue évolutif, qui n’ont pas transité par le WIV. Néanmoins, des analyses basées sur l’« horloge moléculaire » — hypothèse selon laquelle les mutations au sein de l’ADN se produisent au même rythme quelle que soit la lignée considérée — indiquent que la divergence entre les lignées BANAL et SARS-CoV-2 est intervenue il y a une dizaine d’années. BANAL ne peut donc pas être considéré comme étant le fameux chaînon manquant évoqué précédemment. Notons cependant qu’il existe des incertitudes autour des estimations des temps de divergence entre lignées et que l’hypothèse d’horloge moléculaire est probablement trop simpliste dans bon nombre de situations. Il est ainsi envisageable que le chaînon manquant soit issu d’une recombinaison entre virus proches du SARS-CoV-2 tels que RaTG13 ou BANAL, et d’un virus bien plus distant mais présentant le motif de clivage par la furine.
Fin de partie ?
L’hypothèse d’une origine naturelle du SARS-CoV-2 a donc gagné du terrain récemment suite à la découverte des souches laotiennes. Par ailleurs, certaines interrogations liées à l’exceptionnelle célérité avec laquelle l’épidémie s’est transformée en pandémie ont été exprimées début 2020. Cette observation concordait en effet avec l’hypothèse d’une manipulation en laboratoire visant à rendre le SARS-CoV-2 « optimal », manipulations compatibles avec certaines des activités de recherche menées par Shi Zhengli. Or la succession de variants apparus naturellement au cours de la pandémie, toujours plus transmissibles que les souches précédentes, a mis à mal cette théorie.
Des zones d’incertitudes sur les origines de la COVID-19 subsistent néanmoins. Ainsi, alors que des souches proches du SARS-CoV-2 ont été découvertes en Thaïlande, au Cambodge, au Laos et dans la province du Yunnan (sud de la Chine), les premiers cas d’infection chez l’homme se sont déclarés bien plus au nord. Il est entendu que les expériences de terrain ciblent de manière privilégiée les régions au sein desquelles des proches cousins du SARS-CoV-2 ont déjà été observés, créant ainsi un biais d’échantillonnage. Néanmoins, si des virus très proches du SARS-CoV-2 circulaient et circulent encore dans de nombreuses régions d’Asie, voire peut-être même au-delà, comment expliquer que d’autres épisodes de zoonoses, aux conséquences peut-être moins dramatiques que pour la COVID-19, n’aient pas vu le jour auparavant ? La densité élevée d’habitants à Wuhan constitue probablement un terrain idéal pour qu’une zoonose soit suivie de multiples transmissions d’homme à homme. L’hypothèse de la transmission à partir d’un animal infecté, présent sur le marché de Wuhan, en provenance d’une région plus au sud reste donc très probable.
Le mystère autour du site de clivage de la protéine de spicule par la furine — le fameux motif PRRAR — persiste cependant. À l’heure de la rédaction de ce document, aucune autre souche de coronavirus proche du SARS-CoV-2 ne présente cette suite d’acides aminés. Le fameux chaînon manquant permettant d’identifier sans ambiguïté les conditions dans lesquelles le virus a été transmis à l’homme, reste donc à découvrir. Dans cette quête, les méthodes bio-informatiques d’analyse de l’ADN visant à comprendre l’évolution, constituent des outils de choix et permettent d’affiner nos hypothèses dans un cadre méthodologique rigoureux.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

Stéphane Guindon
Chercheur CNRS au Laboratoire d’Informatique, de Robotique et de Microélectronique de Montpellier (LIRMM).
Céline Scornavacca
Directrice de recherche CNRS, à l'Institut des Sciences de l’Evolution de Montpellier (ISEM).