À la recherche de régions codantes

Cette roue visualise le code génétique, c’est-à-dire la correspondance entre les 64 triplets de nucléotides (sur les trois pistes internes) et les 20 acides aminés (sur la piste périphérique).
Par exemple, le triplet ACA code l’acide aminé thréonine, désigné par la lettre T.
Stricto sensu, le génome d’un organisme est l’ensemble de ses gènes ; autrement dit, l’information nécessaire à ses cellules pour synthétiser les protéines qui assurent des fonctions diverses : structure, transport, catalyse, etc. Par extension, le terme génome désigne également le support physique de cette information, la molécule d’ADN (acide désoxyribonucléique), composant des chromosomes présents au sein de chacune des cellules de l’organisme. L’ADN est un enchaînement de nucléotides de quatre types différents distingués par leur base azotée : adénine, thymine, cytosine et guanine, et notés par les initiales A, T, C et G. Et c’est cet enchaînement qui code l’information génétique, au même titre qu’une suite de 0 et de 1 peut coder un son, une image ou une suite d’instructions.
Au sein d’un gène, et plus précisément au sein de sa région codante (ou CDS pour CoDing Sequence), la suite des triplets de nucléotides, appelés codons, dicte la séquence en acides aminés de la protéine.
La correspondance entre les 64 (43) codons possibles et les 20 acides aminés constitue le code génétique, identique à peu de variantes près chez tous les organismes vivants.
Les termes « gène » et « région codante » ne désignent pas exactement les mêmes entités biologiques. Au sein d’un gène, la région codante (souvent désignée par le sigle anglais CDS pour CoDing Sequence) porte la séquence de nucléotides qui, à travers les processus de transcription et de traduction, dicte la séquence en acides aminés de la protéine. Outre la région codante, un gène comporte de plus courtes portions d’ADN qui jouent un rôle dans les processus de transcription et de traduction : promoteur, opérateur, terminateur, RBS, etc.
Des milliards de lettres
Séquencer un génome, c’est déterminer cet enchaînement de nucléotides le long de la molécule d’ADN et le restituer sous la forme d’un texte écrit dans l’alphabet des quatre lettres A, T, C et G. Cette opération, de nature physicochimique, est désormais très largement automatisée et son coût a de ce fait fortement décru ces dernières années. Le premier génome entièrement séquencé, en 1995, celui de la bactérie Haemophilus influenzae, comportait moins de deux millions de nucléotides. Depuis, la séquence de près de 10000 génomes a été déterminée et plus de 30000 brouillons de génomes sont disponibles. Les génomes des eucaryotes, par exemple ceux de l’Homme ou du chien, peuvent comporter plusieurs milliards de nucléotides.
L’obtention du texte d’un génome ne constitue donc plus un objectif scientifique en tant que tel, mais marque le début d’une longue et délicate phase d’analyse. Il s’agit en effet d’identifier, dans ces longs textes dénués de toute « espace » ou « ponctuation », les régions d’intérêt biologique, et plus particulièrement les régions codantes. Cette analyse fait largement appel à l’informatique. La séquence de caractères est en effet un type d’objet bien connu des informaticiens, pour lequel ils ont développé un vaste ensemble d’algorithmes qui continue d’être enrichi par une intense activité de recherche.
Des débuts et des fins qui n’en sont pas
Les biologistes savent que le début d’une région codante dans un génome bactérien est marqué par un triplet ATG, appelé start, et sa fin par l’un des triplets TAA, TAG ou TGA, appelés stop. Le problème de la détection pourrait alors apparaître fort simple : ne suffirait-il pas d’écrire un petit programme qui recherche les occurrences de ces triplets dans la chaîne de caractères qu’est la séquence génomique bactérienne pour délimiter toutes les régions codantes ? La situation se révèle en fait un peu plus compliquée.
En effet, une région codante est une succession non chevauchante de groupes de trois nucléotides, les codons. Elle débute par un codon start et se finit par un codon stop. Il s’agit alors de ne retenir que les couples où le start et le stop sont en phase, c’est-à-dire séparés par un nombre de nucléotides multiple de trois, afin que la région délimitée puisse être considérée comme une succession de codons. Or, il existe trois manières différentes de grouper les éléments d’une séquence trois par trois, selon que l’on commence au premier, au deuxième ou au troisième élément de la séquence. Ces trois manières de grouper les éléments trois par trois déterminent trois phases sur la séquence.
Les régions codantes doivent donc être recherchées dans chacune de ces trois phases, comme si ces dernières déterminaient trois séquences différentes. De plus, un gène peut être porté par n’importe lequel des deux brins complémentaires de cette fameuse double hélice d’ADN. Chacun des brins pouvant être lu selon trois phases, c’est donc en fait dans six séquences différentes, dont cinq virtuelles, que la recherche doit s’effectuer.

Phases de lecture d’une séquence d’ADN.
La séquence « ACCGTAAGACTTGCAC » peut être lue, de gauche à droite, de trois manières différentes selon que l’on commence à considérer les lettres par groupes de trois à la première, à la deuxième ou à la troisième lettre. Ces groupes apparaissent sur la figure alternativement en bleu et en rouge. Il en est de même de la séquence complémentaire, qui est lue de droite à gauche. Une séquence de nucléotides est lue dans le sens 5’ vers 3’. Les notations 5’ et 3’ font référence aux atomes de carbone qui apparaissent aux extrémités libres d’un brin d’ADN.
Mais la vraie difficulté provient de l’occurrence de nombreux triplets stop et start à l’extérieur de régions codantes. Pire encore, des triplets start peuvent se situer à l’intérieur même d’une région codante : ils codent alors la présence de l’acide aminé méthionine dans la protéine.
Les bio-informaticiens ont donc recours à une heuristique qui consiste tout d’abord à rechercher les régions situées entre deux triplets stop en phase et suffisamment longues pour coder une protéine, soit plus de 300 nucléotides. Ils retiennent ensuite le triplet start en phase qui maximise la longueur de la région codante. Bien entendu, la CDS ainsi prédite n’est qu’hypothétique. Le principe est d’accumuler des indices qui confortent cette hypothèse, en particulier la présence de configurations spécifiques de nucléotides, autrement dit de motifs de lettres sur la séquence. Par exemple, l’occurrence, un peu en amont du triplet start, d’un motif associé à ce que les biologistes connaissent pour être un site de fixation du ribosome augmente très sensiblement la pertinence de l’hypothèse.
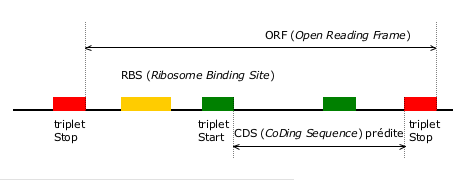
Heuristique d’identification de régions codantes dans une séquence de génome bactérien.
Cette heuristique consiste tout d’abord à rechercher les régions situées entre deux triplets stop en phase (ORF), puis à retenir le triplet start, lui aussi en phase et conférant la longueur maximale à la région codante (CDS). La prédiction est confortée si des motifs biologiques, tels qu’un site de fixation du ribosome (RBS), sont détectés aux endroits attendus.
Enfin, un test assez déterminant consiste à rechercher, dans les bases de séquences déjà annotées, s’il existe, dans le génome d’autres organismes, des séquences similaires à celle de la CDS prédite et connues pour être codantes. Bien évidemment, la pertinence de ce test est totalement dépendante de la qualité des annotations déposées dans les bases de séquences.
Cette démarche assez simple produit cependant des résultats tout à fait corrects sur des séquences de génomes bactériens. Nous vous proposons de le vérifier avec une animation qui détaille cette recherche sur une sous-séquence du génome de la bactérie B. subtilis.
Cette animation vous permet de chercher, dans une sous-séquence du génome de la bactérie B. subtilis, les gènes, ou plus précisément les régions codantes, qui s’y trouvent, et de caractériser la protéine codée par chacun de ces gènes en exploitant la base de protéines UniProt.
Ce processus d’« annotation » est décomposé ici en quatre étapes, listées dans le cadre « Etapes » en haut à gauche de la fenêtre de l’animation.
Attention, lorsque vous quittez, votre recherche ne sera pas conservée.
Pour en savoir plus sur les différentes étapes, voir ci-dessous.
La portion de séquence du génome de B. subtilis s’étend de la position 285 000 à la position 291 000 de la séquence génomique complète, telle qu’elle peut être trouvée dans la base de séquences EMBL ; elle est longue de 6001 nucléotides. Notez que la séquence génomique complète de cette bactérie comporte 4,2 millions de nucléotides et contient 4106 gènes.
La « carte génomique » affichée par l’applet fait apparaître les triplets start sous la forme de traits verts verticaux et les triplets stop sous la forme de traits rouges, dans les trois phases numérotées +1, +2 et +3 sur le brin direct (orienté de la gauche vers la droite) et dans les trois phases numérotées –1, –2 et –3 sur le brin complémentaire (orienté de la droite vers la gauche).
Au sein d’un gène, un RBS ne fait pas partie de la région codante, il n’apparaît donc pas obligatoirement dans la même phase que les start et stop. C’est pourquoi les motifs RBS apparaissent sur deux lignes distinctes, l’une associée au brin direct, l’autre au brin complémentaire.
En cliquant sur les boutons étiquetés « START », « STOP » et « RBS », vous faites disparaître et apparaître respectivement les triplets start et stop et les motifs RBS. Ainsi, en ne faisant apparaître que les triplets stop, plusieurs ORF suffisamment longues se distinguent assez nettement comme des « trous » sur chaque ligne associée à une phase. C’est au sein de ces ORF que vous sélectionnerez une première CDS candidate.
Bien que les RBS apparaissent sur la carte génomique, il vous est conseillé de ne pas en tenir compte pour la sélection de CDS et de vous en tenir aux critères énoncés dans le texte : choix sur une phase d’une ORF de plus de 300 nucléotides et du start immédiatement en aval du codon stop qui précède l’ORF ; le codon stop de la CDS est le codon stop qui termine l’ORF.
Vous devez donc choisir un start et un stop dans une même phase. Le choix se fait en positionnant la flèche de la souris sur le trait correspondant au triplet start ou stop. Si le positionnement est correct, la flèche se transforme en une main à l’index pointé. En cliquant, vous sélectionnez le triplet. La sélection d’un triplet start (respectivement stop) annule l’éventuelle sélection précédente d’un autre triplet start (respectivement un autre triplet stop).
La fenêtre « Messages » fait apparaître des messages qui sont susceptibles de vous aider en cas de choix erronés. Par exemple, un message vous rappellera que, sur les phases négatives, le triplet start doit se situer « à droite » du triplet stop.
Enfin, il est souvent nécessaire d’effectuer une opération de zoom pour mieux distinguer les triplets et les sélectionner. À un niveau de zoom très élevé, vous verrez apparaître les lettres qui composent la séquence de chaque brin.
Dans cette deuxième étape, la CDS candidate est traduite en une séquence polypeptidique, c’est-à-dire un enchaînement d’acides aminés. À chaque codon est associé, via le code génétique, un acide aminé. La séquence de la CDS est ainsi parcourue séquentiellement du premier codon, un start, au dernier codon, un stop.
Ici, le code génétique est figuré par une roue portant des disques concentriques découpés en secteurs : le premier acide nucléique du codon détermine l’un des quadrants du disque le plus intérieur, le deuxième l’un des 16 secteurs figurant dans le disque immédiatement supérieur et le troisième l’un des 64 secteurs en périphérie du secteur précédent. Ces quadrants et secteurs prennent la même couleur rosée pour un triplet lu sur la séquence. Sur le dernier disque figurent les acides aminés.
En cliquant sur le secteur associé à un acide aminé, vous obtenez sa structure chimique, son nom complet et son code en une et trois lettres. Par exemple, à la lecture du codon CTG, c’est le secteur correspondant à l’acide aminé Leucine qui est sélectionné ; cet acide aminé peut être désigné par la seule lettre L ou par les trois lettres Leu.
Le résultat de l’application de l’algorithme de traduction de la séquence de la CDS que vous avez sélectionnée est une séquence de lettres écrite dans l’alphabet des 20 lettres associées aux 20 acides aminés.
Vous devrez tout d’abord utiliser la roue pour traduire 5 triplets en cliquant sur les lettres qui les composent. Vous pourrez ensuite utiliser le menu pour traduire successivement les autres triplets, ou aller directement à la fin de la traduction. Si N est la longueur de la CDS candidate, la longueur de la séquence polypeptidique est N / 3 – 1 ; en effet, le stop n’est pas traduit.
La traduction formelle de la séquence de la CDS candidate en une séquence polypeptidique est bien entendu une abstraction et une simplification de la réalité biologique.
Au sein de la cellule, la CDS est une région d’un des deux brins de la molécule d’ADN. Cette région est tout d’abord copiée en une molécule d’ARN, identique à la région d’ADN originelle, à la seule différence que l’uracile (désignée par la lettre U) y remplace la thymine (T). Une séquence d’ARN se représente donc par une chaîne de caractères écrite dans l’alphabet A, U, C et G. Ce processus de copie préserve l’archive qu’est la molécule d’ADN ; il est appelé « transcription ». La transcription fait intervenir une molécule, l’ARN polymérase, qui parcourt la région d’ADN et synthétise progressivement la copie.
La molécule d’ARN ainsi obtenue est ensuite traduite en une chaîne polypeptidique. Cette traduction fait intervenir des molécules diverses, telles que les ribosomes et les ARN de transfert. Un ARN de transfert est une molécule qui réalise la correspondance entre codon et acide aminé : elle porte d’un côté un anticodon, qui peut s’apparier à un codon lu par le ribosome, et de l’autre l’acide aminé correspondant à ce codon. Autrement dit, l’ensemble des différents ARN de transfert constitue la matérialisation du code génétique.
La séquence polypeptidique qui résulte de l’étape précédente peut être comparée aux séquences des 185 000 protéines répertoriées dans la base de données UniProt. Le principe est de trouver des séquences qui ressemblent à la vôtre. L’existence de séquences similaires signifie que votre protéine ressemble à une protéine répertoriée, qui appartient à un organisme plus ou moins apparenté.
Ici, l’organisme est la bactérie B. subtilis dont le protéome, c’est-à-dire l’ensemble des protéines codées par les gènes, est bien connu. Si vous avez sélectionné une vraie CDS, votre protéine doit ressembler à une des protéines connues du protéome de B. subtilis. Inversement, si vous avez sélectionné une région qui n’est pas une CDS, la recherche dans UniProt ne ramènera pas de protéine dont la séquence ressemble à votre séquence.
Bien entendu, lors de l’identification de régions codantes dans un génome nouvellement séquencé, les recherches dans des bases de données renvoient des séquences similaires exclusivement associées à d’autres organismes que celui qui est étudié.
Pour lancer la recherche, il vous faut tout d’abord « copier » la séquence traduite qui apparaît dans le cadre intitulé « Séquence polypeptidique », puis lancer le programme de recherche dans la base UniProt.
Une nouvelle fenêtre de votre navigateur s’ouvre. Il vous suffit alors de « coller » la séquence polypeptidique dans la zone de saisie, puis de cliquer sur le bouton « Run Blast » pour lancer la recherche (qui met en œuvre un programme appelé « Blast »).
La recherche prend normalement quelques secondes, mais peut être plus longue si le serveur est très sollicité (vous n’êtes probablement pas le seul à effectuer une recherche dans UniProt en cet intant).
Le programme d’interrogation de la base UniProt renvoie une liste de protéines dont la séquence ressemble à la vôtre, même si les deux séquences n’ont pas la même longueur. Il affiche un degré de similarité qu’il n’est pas nécessaire ici d’interpréter de façon précise. Il vous suffit de regarder le code couleur des alignements : une protéine avec le code de couleur rouge est très semblable à la séquence donnée en entrée ; un code bleu indique une protéine trop peu semblable.
Si la première ligne du résumé affiche une icône avec une étoile, vous pouvez considérer que la recherche confirme votre CDS candidate. Faites alors un « copier » du nom de la protéine qui apparaît dans la colonne « Entry name » (ou encore du libellé de la protéine qui apparaît en gras sur la ligne en dessous), retournez dans la fenêtre de l’applet et collez le nom ou le libellé dans le cadre en bas à droite de la fenêtre, puis cliquez sur le bouton « Valider l’annotation ». Vous passez alors à la quatrième et dernière étape.
Dans le cas contraire, cliquez sur le bouton « Annuler l’annotation », vous revenez alors à la première étape pour choisir une autre CDS candidate.
La carte fait maintenant apparaître une épaisse flèche bleue en coïncidence avec la zone que vous aviez initialement sélectionnée comme une CDS candidate. Au-dessus de cette flèche apparaît le nom ou le libellé de la protéine retrouvée similaire dans UniProt.
Vous pouvez alors recommencer ce processus d’identification et de validation d’une région codante dans cette portion du génome de la bactérie B. subtilis.
Notez bien que des régions codantes ne peuvent se chevaucher, qu’elles soient sur un même brin ou non, qu’elles soient sur une même phase ou non.
Animation HTML5/JS réalisée par Hugo Lehmann, Centrale Lille Projets, adaptée d’une applet réalisée par Régis Monte et Guillaume Surrel, à partir d’un programme Java initialement développé par Gaël Faroux puis amélioré par Delphine Baratin.
Des archipels perdus dans un océan de lettres
Dans les séquences de génomes eucaryotes, la recherche de régions codantes est malheureusement beaucoup plus délicate, et ce pour deux grandes raisons. Tout d’abord, les gènes eucaryotes sont séparés par de très longues régions, dites intergéniques. Ainsi, les gènes occupent-ils moins de 30 % de la séquence humaine. Ensuite, un gène eucaryote comporte une succession de séquences codantes, appelées exons, et non codantes, appelées introns. De ce fait, au bout du compte, les régions codantes occupent moins de 3 % de la séquence du génome humain. Cette structure morcelée complique évidemment le problème de la prédiction de la région codante, puisqu’il ne suffit plus de déterminer un début et une fin, mais également les différentes frontières exon-intron.
Les bio-informaticiens sont donc amenés à affiner leur démarche d’analyse. Un de leurs outils favoris met en œuvre des modèles probabilistes, dits de Markov, qui, convenablement configurés, sont capables de détecter les différences de « style » entre régions non codantes et codantes. D’autres méthodes tenteront de détecter les motifs plus ou moins bien définis qui correspondent aux jonctions exon-intron. Là encore, c’est la superposition et la confrontation des résultats produits par plusieurs méthodes, éventuellement concurrentes, qui permettront de conforter et d’affiner les prédictions.
Les méthodes d’analyse de génomes actuelles sont largement perfectibles, en particulier lorsqu’elles s’appliquent aux séquences eucaryotes : certains gènes ne sont pas décelés, d’autres sont indûment prédits, les débuts et fins de régions codantes peuvent être erronés. Mais quels que soient les progrès méthodologiques, les résultats des méthodes bio-informatiques conserveront le statut de prédictions et devront toujours être validés par une démarche expérimentale. Tout l’intérêt du recours à la bio-informatique est bien entendu de réduire considérablement le nombre d’hypothèses à tester et d’accélérer ainsi le processus de transformation des données en connaissances.
Une première version de ce document est parue dans le Gluon, édité par l’université Joseph Fourier de Grenoble, dans son numéro de mars 2006. Première publication sur Interstices en septembre 2006. Mise à jour en avril 2019 avec la contribution de Pierre Peterlongo.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !
