Accélérer l’exploration et l’analyse des textes des génomes
Le séquençage des génomes
Depuis 2001, nous disposons du texte complet du génome humain. C’est la traduction fidèle de l’immense molécule d’ADN présente au cœur de chacune de nos cellules. De la même manière, des centaines d’autres organismes sont en cours de traduction, car les techniques de séquençage sont maintenant bien maîtrisées, et pour des coûts toujours plus réduits. Pratiquement, depuis que l’on sait lire le texte des génomes, le volume d’information double chaque année.

Séquenceur automatique multi-capillaire
(Plate-forme de séquençage de la Station Biologique de Roscoff).
© CNRS Photothèque – Richard Lamoureux
D’un point de vue informatique, le séquençage se traduit par la production d’un texte dont l’alphabet se réduit à 4 caractères : A, T, G et C. Chacun représente un des 4 nucléotides inscrit sur un des brins de la double hélice d’ADN : l’adénine (A), la cytosine (C), la guanine (G) et la tymine (T). La taille des textes varie énormément d’un organisme à un autre. L’homme possède un génome correspondant à un texte de 3 milliards de caractères (1 000 livres de 1 000 pages). Le génome d’une bactérie ne représente lui que quelques millions de caractères (un ou deux livres de 1 000 pages).
Comprendre le texte des génomes
Si transcrire une information biochimique (l’ADN) en une longue suite de caractères (le texte des génomes) est un problème résolu, en comprendre la signification est, par contre, un problème d’une tout autre complexité. L’élément de base est le gène. Il correspond à une courte séquence de caractères localisée à un endroit précis dans le texte du génome. La cellule sait reconnaître ces séquences et les traduire en protéines, véritables briques de base dont est constitué tout être vivant. Dans ces zones, un simple caractère erroné peut conduire à une mauvaise traduction de la protéine. Celle-ci, mal formée, ne remplit alors pas (ou mal) son rôle. Elle peut alors entraîner un dysfonctionnement des cellules où ce gène s’exprime et être à l’origine de maladies plus ou moins graves.
Pour analyser les génomes à partir de leur texte, les biologistes disposent d’une palette d’outils informatiques conséquente. La plupart d’entre eux ont été développés dans des centres de recherche publics et sont disponibles gratuitement. De manière non exhaustive, ils permettent d’établir des phylogénies, de localiser des gènes, de détecter des structures génomiques remarquables, etc. Mais la tâche qui se situe probablement en toute première position est l’exploration des banques de séquences publiques.
Une tâche de base : explorer les banques
La banque américaine GenBank, la banque japonaise DDBJ et la banque européene EMBL mettent en commun toutes les séquences d’ADN du domaine public séquencées de par le monde, au sein de l’INSDC (International Nucleotide Sequence Database Collaboration). Les séquences sont annotées par les chercheurs eux-mêmes et les banques sont mises à jour quotidiennement. Elles reflètent donc l’état des connaissances le plus récent de la communauté scientifique. Ainsi, un chercheur ayant, par exemple, localisé un gène dont la fonction lui est inconnue, peut le comparer aux centaines de millions de gènes déjà emmagasinés dans ces banques. S’il trouve des ressemblances textuelles avec d’autres gènes dont on connaît la fonction, il peut alors bâtir plus rapidement des hypothèses sur les fonctionnalités de ce nouveau gène. En effet, deux gènes présentant des similarités significatives au niveau du texte conduisent en général à des protéines de forme similaire. La forme induisant la fonction, on peut en conclure qu’une similarité textuelle peut être révélatrice d’une similarité fonctionnelle.
Mais cette recherche a un coût. Il faut analyser systématiquement les centaines de millions de séquences des banques. Des programmes très efficaces, tels que BLAST par exemple, ont été développés pour limiter au maximum les temps de recherche.
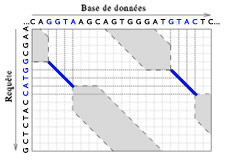
Représentation de l’espace de recherche de similarités entre une séquence requête et une base de données.
Sur cet exemple, deux ancrages ont été trouvés : GGTA et GTAC. Il ne reste donc à parcourir que les parties grisées de part et d’autre de ces zones. Le gain en temps d’exécution est le rapport de la surface totale sur la surface grisée.
BLAST (Basic Local Alignment Search Tool) est probablement le programme le plus utilisé en biologie moléculaire. Sa rapidité provient du fait qu’il réduit considérablement l’espace de recherche en se focalisant uniquement sur des régions potentiellement intéressantes.
Dans BLAST, une requête est une séquence dans laquelle on cherche à mettre en évidence des zones de similarité avec toutes les séquences de la banque qu’on interroge. Trouver des similarités entre séquences revient à mettre en correspondance un maximum de caractères identiques consécutifs entre deux textes distincts.
Pour trouver rapidement les alignements, le logiciel BLAST fait l’hypothèse qu’ils se situent dans des régions où les textes ont au moins en commun un mot de K caractères. Par défaut, on s’intéresse aux régions qui partagent au moins des mots de 11 caractères.
Cette méthode de recherche de similarité est donc relativement rapide car elle se concentre immédiatement sur un petit nombre de zones privilégiées.
Elle présente cependant un inconvénient : le programme BLAST peut ne pas détecter tous les alignements biologiquement significatifs. On peut bien sûr abaisser le seuil de détection en choisissant de faire fonctionner BLAST avec des mots plus courts (par exemple 7 caractères) pour augmenter la sensibilité. Le prix à payer est alors un temps de calcul plus long.
Mais l’explosion des données génomiques force à trouver d’autres solutions, pour que ce temps reste raisonnable. En effet, le rythme de croissance des données génomiques est supérieur à l’évolution des performances des ordinateurs. Dans un cas, le volume double tous les 12 mois alors que dans l’autre cas, d’après la loi de Moore, les performances doublent (seulement !) tous les 18 mois. L’écart ne cesse de se creuser, ce qui implique des temps de traitement de plus en plus longs.
Un temps de réponse le plus court possible
Pour garantir une interactivité raisonnable (un temps de réponse de quelques minutes, voire quelques dizaine de secondes maximum), plusieurs approches sont possibles. La première, mise en œuvre sur les serveurs web des centres de calcul bio-informatique, utilise des supercalculateurs, capables de traiter des dizaines de milliers de requêtes chaque jour. La seconde, plus récente, utilise des clusters de dizaines ou de centaines de PC standard. Ces deux solutions sont très efficaces. Les performances dépendent directement du nombre d’ordinateurs : plus il est important, plus le calcul est fait rapidement.
En revanche, ce sont des solutions coûteuses que seules des structures d’une certaine importance peuvent s’offrir. La mise à disposition de serveurs web publics ne résout pas forcément le problème : les données transitent par Internet et peuvent être interceptées, ce que certains laboratoires (publics ou privés), pour des raisons économiques et/ou de confidentialité, ne peuvent tolérer.
Des machines spécialisées
Conception de la carte RDISK dédiée à l’exploration des banques de données.
© INRIA – projet SYMBIOSE
Une autre alternative, développée dans l’équipe de bio-informatique de l’IRISA, est la conception de machines dédiées à l’exploration des banques génomiques. Le but est de proposer des systèmes très rapides, basés sur d’autres architectures que celles des microprocesseurs. Ces dernières, architectures généralistes, ont l’avantage de supporter n’importe quel traitement. Par contre, malgré leur vitesse de fonctionnement importante (quelques Gigahertz), leurs performances peuvent être bridées par le contexte applicatif. Les machines spécialisées y remédient, mais au prix d’une perte de généralité importante. Pour prendre une analogie avec le monde automobile, les machines spécialisées représentent les Formule 1 ; elles sont optimisées dans un but précis : gagner des courses sur un circuit. En revanche, on ne peut y atteler une caravane et partir en vacances !
L’interrogation des banques génomiques est avant tout conditionnée par l’accès rapide aux données. On ne pourra pas aller plus vite que le temps qu’il faut pour les lire sur les disques et ce, quel que soit le dispositif de traitement en aval du support de stockage. Inversement, il ne faut pas que la lecture des données soit ralentie par un mécanisme incapable d’absorber le flux en provenance des disques. L’idéal est une architecture équilibrée, à la fois capable de lire le texte des génomes à toute vitesse et capable d’effectuer, sur ces textes, des traitement à la volée, c’est à dire au fur et à mesure de la lecture des séquences génomiques.
Un prototype parallèle et reconfigurable
Le prototype réalisé dans notre équipe met en pratique cette réflexion. L’accès rapide aux données est assuré en répartissant le texte des génomes sur une batterie de 48 disques qui sont lus simultanément. Ainsi, on divise par 48 le temps de lecture. Mais surtout, un dispositif matériel particulier est attaché à chaque disque. Il a pour tâche de filtrer les séquences génomiques pour ne laisser passer que celles qui présentent un intérêt potentiel par rapport à une interrogation précise de la banque.

Prototype de la carte RDISK dédiée à l’exploration des banques de données.
© INRIA – Richard Lamoureux
Le rôle du filtre est primordial : il doit à la fois être très rapide pour ne pas freiner les disques et très sélectif pour ne retenir que les quelques séquences pertinentes parmi les millions emmagasinées dans les banques. Ce filtrage est supporté par un composant FPGA qui, à l’opposé d’un microprocesseur, ne déroule pas un programme, mais câble directement ce programme en portes électroniques élémentaires. La différence est qu’un programme qui demande l’exécution de plusieurs micro-instructions sur un microprocesseur est ici réalisé en une seule micro-instruction spécialisée. Celle-ci peut être modifiée à la demande : il s’agit alors de reconfigurer le composant FPGA, c’est-à-dire de lui indiquer par le biais d’un nouveau plan de câblage des portes électroniques la nouvelle micro-instruction ou, dans notre cas, la nouvelle fonction de filtrage.
Des traitements qui prendraient plusieurs jours sur un ordinateur récent peuvent ainsi être effectués par notre prototype en quelques dizaines de minutes. Les séquences retenues par le dispositif de filtrage peuvent alors être analysées efficacement par un ordinateur standard.
Chez le chien, les gènes impliqués dans l’odorat se caractérisent par un ensemble de 5 motifs particuliers plus ou moins présents dans chacun d’eux. Un gène peut en contenir 5, alors qu’un autre n’en possède que 2 ou 3. Notre prototype a été mis à contribution pour localiser et comptabiliser ces gènes avant que le texte du génome ne soit entièrement disponible. Nous avons travaillé directement sur les données brutes de séquençage, soit 36 millions de courtes séquences d’ADN. Sur chaque séquence, il fallait tester la présence ou non des 5 motifs, en autorisant, de plus, un certain nombre d’erreurs.
Sur un ordinateur récent, le traitement prend plusieurs jours. Sur notre prototype, les 5 motifs ont été câblés pour sélectionner les séquences d’ADN qui présentaient un ou plusieurs de ces motifs. Le traitement ne dure qu’une quinzaine de minutes et ne retient qu’environ 2 % des séquences. Ce sous-ensemble, qui représente un volume d’information tout à fait gérable efficacement par un ordinateur standard, a ensuite été analysé et a permis d’identifier 1 120 gènes.
Filtrage de bases de données sur le prototype RDISK
S. Guyétant, D. Lavenier, SympAAA’2003 9e Symposium en Architectures de Machines et Adéquation Algorithme Architecture, La Colle sur Loup, 2003.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

Dominique Lavenier
Directeur de recherche CNRS à l’Institut de recherche en informatique et systèmes aléatoires (IRISA), membre de l'équipe de recherche GENSCALE.