La bio-informatique en protéomique : analyse des spectres de masse
C’est en 1994 que le terme « protéome » a été employé pour la première fois, afin de désigner le pendant protéique d’un génome. Il s’agit de l’ensemble des protéines produites à partir d’un génome à un moment et dans des conditions données.
1. La cellule : du génome au protéome
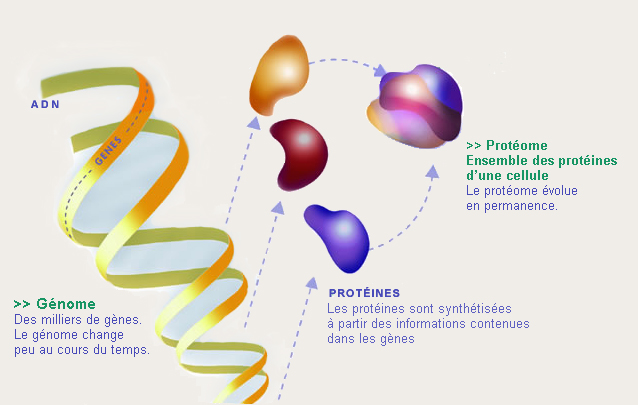
La protéomique, l’étude du protéome et de ses évolutions, fournit des informations précieuses sur la façon dont la cellule réagit à son environnement (médicaments, hormones, température, etc.).
(image ORNL – U.S. Department of Energy Genomics : GTL Program)
De quoi donner le vertige : on estime que les quelque 30 000 gènes humains seraient à l’origine d’au moins un million de protéines. Ce facteur multiplicatif entre nombre de gènes et de protéines existe pour tous les organismes, en particulier eucaryotes, car un gène contient le plus souvent l’information qui permettra de fabriquer plusieurs protéines différentes. Plutôt que de s’attaquer de front à cette montagne, les chercheurs ont choisi de la découper en morceaux. Ils ont adopté une approche d’analyse différentielle : pour un type cellulaire donné, ils suivent les variations du protéome causées par diverses modifications de son environnement (stimulus, état pathologique…). C’est malgré tout par milliers que les protéines sont présentes dans les échantillons biologiques d’intérêt, et leur identification est l’étape clé préalable à toute autre étude.
2. La protéomique à haut débit

Spectromètre de masse quadripolaire triple Pe-Sciex Api 365
© Institut Pasteur.
Emboîtant le pas aux ambitieux programmes de séquençage de génome, l’étude systématique des protéines a pu être envisagée grâce, en particulier, au développement de la spectrométrie de masse. Cela fait environ trente ans que cette technologie s’est adaptée aux exigences des biologistes, mais il a fallu attendre le milieu des années quatre-vingt-dix pour que, couplée aux outils bio-informatiques, elle permette une réelle avancée de l’analyse protéomique.
Deux difficultés, inhérentes à la composition cellulaire, sont rencontrées par les scientifiques : les protéines à caractériser sont très nombreuses, et certaines sont présentes en très faible quantité. En général, les biologistes cherchent à traiter le second problème en amont des étapes de caractérisation, en tentant par différents moyens d’augmenter dans l’échantillon étudié les proportions des protéines peu abondantes. Mais pour faire face au nombre et à la diversité des protéines, c’est au niveau des performances des techniques d’analyse employées que tout se joue.
Principe des techniques biologiques employées
La caractérisation débute le plus souvent par une séparation des différentes protéines du mélange étudié.
Gel d’électrophorèse bidimensionnel (image CEA).
Plusieurs techniques peuvent être employées, on peut citer l’électrophorèse bidimensionnelle sur gel ou la chromatographie liquide. Chaque protéine ainsi purifiée subit un traitement enzymatique afin d’être découpée en peptides. Les enzymes qui digèrent la protéine la coupent à des endroits précis, aussi une protéine donnée sera-t-elle toujours fragmentée de la même façon. C’est ce mélange de peptides qui est alors analysé par spectrométrie de masse (MS). Pour donner une idée de la sensibilité de la technique, précisons que le microlitre d’échantillon injecté dans le spectromètre à chaque essai contient des quantités de protéine digérée de l’ordre du 1/10 voire 1/100 de microgramme.
Le spectre obtenu reflète le « profil de digestion » de la protéine par l’enzyme utilisée (le plus souvent la trypsine) : c’est une sorte d’empreinte de la protéine.
L’analyse peut s’arrêter là. Mais il arrive que le spectre obtenu par MS soit ambigu, c’est-à-dire qu’il ne soit pas assez spécifique et puisse correspondre à plusieurs protéines. On emploie alors une autre technique de spectrométrie de masse, dite « en tandem » (ou MS/MS), dans laquelle chacun des peptides est ensuite lui-même fragmenté, et ses produits de fragmentation analysés. Cette technique apporte des informations concernant la séquence de la protéine étudiée, car la fragmentation des peptides a lieu sur les liaisons chimiques entre les acides aminés et les « libère » de la chaîne peptidique, un par un ou en sous-fragments très courts.
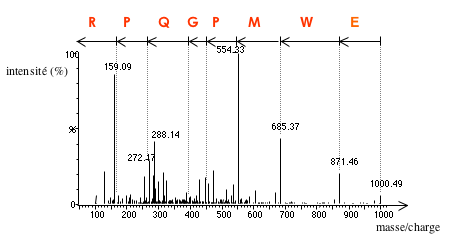
Exemple de spectre MS/MS.
En abscisse, la masse des ions (plus exactement, le rapport masse/charge) ; en ordonnée, le pourcentage des ions possédant une masse donnée. En pratique, seul l’espacement entre les pics est interprété, pas leur hauteur. En interprétant ces espacements, il est possible de reconstituer la séquence peptidique. Sur cet exemple, la lecture du spectre de droite à gauche permet de reconstituer la séquence EWMPGQPR (chacun des 20 acides aminés est ici désigné par un code d’une lettre).
Les spectromètres de masse actuels permettent d’enregistrer, de manière entièrement automatique, plusieurs milliers de spectres MS/MS en quelques heures. C’est leur interprétation qui pose alors problème. Le rythme d’obtention de cette masse de données dépasse bien sûr largement celui d’une analyse « manuelle ». Quant au dépouillement automatique des spectres, il a encore beaucoup de progrès à faire.
Identification automatique des protéines
Cette identification automatique fait appel à la bio-informatique et au criblage des banques de données.
Une approche directe consiste à comparer le spectre de la protéine inconnue aux spectres virtuels de toutes les protéines d’une banque de données. Ces spectres virtuels sont obtenus grâce à des logiciels capables, d’une part, de simuler la digestion enzymatique de n’importe quelle protéine, puis de construire le spectre de masse que donnerait chacun des fragments peptidiques. L’outil est très puissant, mais limite l’identification à des protéines déjà répertoriées dans les banques.
Une autre stratégie, plus « souple », inclut une étape préalable d’interprétation du spectre expérimental en séquence d’acides aminés. C’est cette séquence potentielle qui est ensuite comparée à celles contenues dans les banques de données. Les logiciels qui recherchent les similarités de séquences ayant été conçus pour accepter des variations (substitution, insertion ou délétion d’acide aminé), l’identification est plus ouverte et adaptée à la question posée. Par contre, le bât blesse au niveau de l’interprétation du spectre, exercice très complexe que maîtrisent les spécialistes en spectrométrie de masse, mais pas encore les programmes informatiques mis au point dans ce but.
L’équipe HELIX de l’INRIA Rhône-Alpes a développé des outils informatiques, basés sur une version améliorée de cette seconde approche, qui permettent de relier directement les résultats de l’analyse protéomique aux séquences du génome correspondant.
3. Des étiquettes peptidiques aux gènes
La procédure d’identification est la suivante : détermination « d’étiquettes peptidiques » ou « PST » (Peptide Sequence Tag), pour chaque spectre (donc pour chaque fragment de protéine), puis criblage des banques de séquences avec ces étiquettes.
Construction des étiquettes peptidiques
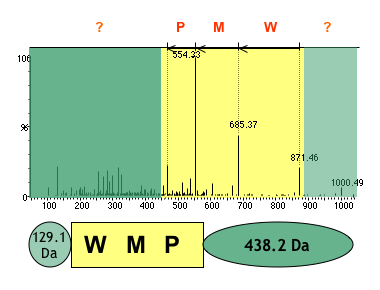
Étiquette peptidique ou PST.
Elle est obtenue en restreignant l’interprétation du spectre à sa zone centrale (en jaune sur la figure), généralement la plus lisible, conduisant ici à la séquence d’acides aminés WMP. Les zones adjacentes (en vert) ne sont décrites que par la masse totale (exprimée ici en Dalton) des acides aminés qui les composent.
L’astuce réside dans la simplification de l’étape d’interprétation des spectres de masse. Au lieu de traiter la totalité du spectre, seule une zone précise et facilement interprétable est prise en compte. Elle correspond à une courte séquence, de trois à cinq acides aminés, située au centre du fragment peptidique analysé. De part et d’autre de cette zone du spectre, seule l’information brute, c’est-à-dire la masse des fragments du mélange, est utilisée. On obtient ainsi des « étiquettes peptidiques », données « hybrides » constituées de deux valeurs de masse encadrant une séquence peptidique.
Un premier logiciel, Taggor, génère à partir d’un spectre MS/MS toutes les étiquettes possibles. Seules sont conservées les plus susceptibles d’être fiables, c’est à dire celles dont la séquence peptidique a été interprétée à partir de la zone la plus informative du spectre. Il reste cependant des étiquettes peptidiques erronées, mais la seconde étape d’identification va permettre d’en faire le tri.
À la recherche des gènes correspondants
C’est là où tout se complique ! Car il s’agit de faire correspondre les étiquettes peptidiques avec les séquences, d’ADN ou de protéines, stockées dans les banques. Avec des séquences protéiques, la recherche est évidente. Pour l’ADN, cela suppose une traduction préalable des séquences en acides aminés. Or, si l’on ne sait pas où se trouvent les gènes, il faut prendre en compte les différentes possibilités de lecture de l’ADN : trois phases de lecture possibles sur chacun des deux brins d’ADN, soit six « textes » à lire pour chaque séquence.

Les 6 phases de lecture possibles.
La traduction d’une séquence génomique en une séquence d’acides aminés, un polypeptide, s’effectue dans la cellule, et plus précisément au niveau des ribosomes, en associant à chaque suite contiguë de trois acides nucléiques, appelée codon, un acide aminé. La correspondance entre les 64 (43) codons et les 20 acides aminés constitue le code génétique. Or, il existe trois façons de grouper les caractères d’une séquence trois par trois, suivant que l’on commence le groupement à la première, la deuxième ou la troisième lettre : on parle de trois phases de lecture de la séquence, notées +1, +2 et +3. De même, il existe trois phases de lecture de la séquence du brin complémentaire, notées -1, -2 et -3, le signe – soulignant l’inversion du sens de lecture de ce brin. Il existe donc six phases de lectures distinctes pour une même séquence génomique. Un gène peut être localisé sur le brin direct ou sur le brin complémentaire, et dans chaque cas, sur l’une des trois phases. En pratique donc, la recherche de gènes s’effectue dans les six phases, autrement dit dans six séquences différentes.
Faire correspondre les étiquettes peptidiques avec les séquences d’ADN génomique, c’est le rôle d’un second logiciel, PepMap, qui extrait des étiquettes peptidiques à partir de spectres MS/MS et les localise sur la séquence d’un génome, puis utilise l’agrégation statistique pour étudier la pertinence des résultats.
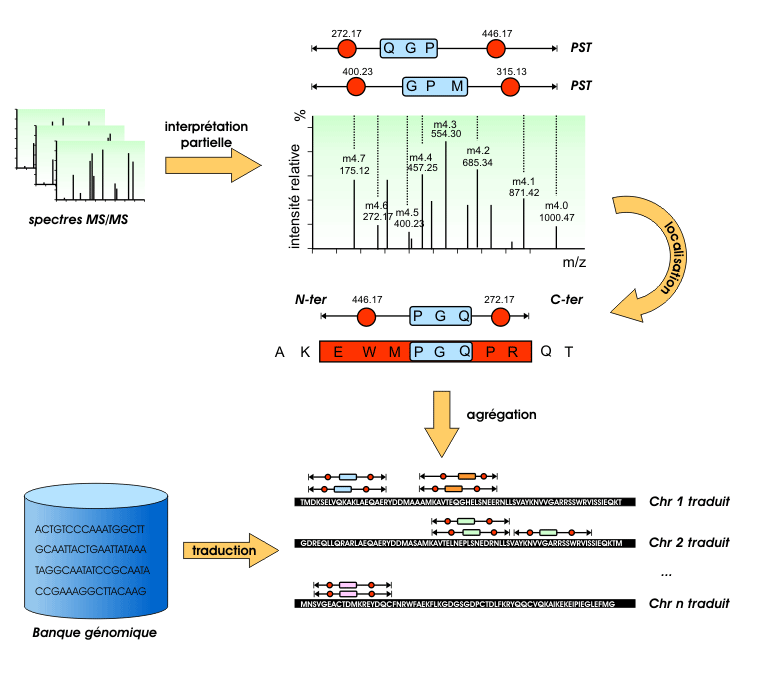
Principe de fonctionnement du logiciel PepMap.
L’algorithme se décompose en trois phases :
1. Conversion d’un spectre MS/MS, associé à un fragment peptidique, en une liste de PST.
2. Localisation des étiquettes peptidiques sur des séquences.
3. Agrégation statistique des occurrences de PST de plusieurs spectres.
PepMap est employé pour chercher le long des séquences d’ADN les séries de 3, 4 ou 5 acides aminés des étiquettes peptidiques produites par Taggor. Des contraintes sont ajoutées à cette recherche par les données de masse flanquant le petit peptide.
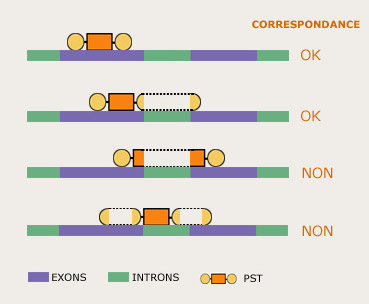
Différentes possibilités de correspondances entre les étiquettes peptidiques et les gènes.
Dans le cas de l’étude d’un organisme eucaryote, il suffit cependant qu’une seule des deux masses soit reconnue, à cause de la structure des gènes mosaïques. En effet, si la séquence codant pour le petit peptide est située à la frontière avec un intron, la séquence du reste du fragment analysé par MS/MS – et donc la masse portée par l’étiquette – va se trouver de l’autre côté de l’intron.
Les bio-informaticiens ont travaillé à la rapidité d’exécution de cette partie de l’analyse, qui pouvait devenir l’étape limitante du processus. En effet : demandez donc à votre logiciel de traitement de texte de rechercher le mot « des » dans un texte : il en trouvera à chaque ligne. Et si vous parcourez le texte pour les lister, cela prendra beaucoup de temps. Le principe est le même ici : la probabilité de trouver une séquence de 3 à 5 acides aminés est élevée, et d’autant plus élevée que la séquence passée en revue est longue – ce qui est le cas pour des génomes entiers.
C’est pourquoi les concepteurs de PepMap ont délaissé une approche de « scan » de toute la séquence étudiée au profit d’une tactique plus astucieuse. Considérons des peptides de 3 acides aminés. Le logiciel construit un tableau contenant l’ensemble des combinaisons possibles de tripeptides – soit 203, puisqu’il y a 2 acides aminés différents. Ensuite, chacun des six « textes » est lu, successivement, par triplets d’acides aminés, en décalant la lecture d’un acide aminé à chaque fois (par exemple, si chaque lettre désigne un acide aminé, la séquence D E F G H donne les tripeptides DEF, EFG et FGH). Sont alors reportées dans les cases correspondantes du tableau les positions des triplets (case DDD : rien ; case DDE : rien ; … ; case DEF : position 1 ; case DEG : rien ; … ; case EFG : position 2 ; etc.). C’est dans ce tableau que PepMap va rechercher si les étiquettes générées par Taggor ont une ou plusieurs occurrences, et à quelle(s) positions(s). Pour chacune de ces positions, le logiciel vérifie alors si la concordance est confirmée par les données de masse de l’étiquette.
Dans un second temps, PepMap va évaluer, entre toutes les occurrences observées, celles qui ont le plus de chance de correspondre effectivement à un gène. Il commence par repérer les zones de la séquence où sont groupées plusieurs étiquettes. De tels « agrégats » peuvent indiquer la présence d’un gène. Afin de le confirmer, le logiciel fait une étude statistique pour calculer si les agrégats se sont formés aléatoirement, ou s’ils sont significatifs. Des « scores d’intérêt » sont attribués à chaque étiquette peptidique : si une étiquette n’existe qu’une fois sur la séquence, son score est maximal car sa présence indique la présence du gène de la protéine analysée. Le score de l’agrégat est la somme des scores des étiquettes qui le composent. PepMap compare ce score avec le score calculé pour une « séquence-mélangée », c’est-à-dire dans laquelle l’ordre des acides aminés de la séquence étudiée aurait été complètement modifié, aléatoirement. L’agrégat est statistiquement pertinent lorsque son score dépasse celui de cette « séquence-mélangée ».
4. Les apports de ces outils informatiques
Identifier les gènes de protéines inconnues est une étape cruciale de l’annotation du génome, étape que les bio-informaticiens tentent d’automatiser par différentes approches. Dans ce but, la confrontation entre spectres MS/MS et séquences génomiques réalisée par les logiciels Taggor et PepMap donne de premiers résultats très encourageants.
La méthode décrite ici possède l’avantage supplémentaire de localiser précisément exons et introns. De plus, elle peut être appliquée non seulement à une protéine isolée, comme nous l’avons décrit, mais aussi à un mélange de protéines. En effet, même si elles sont « construites » à partir de spectres MS/MS de fragments provenant de plusieurs protéines, les étiquettes peptidiques vont être en quelque sorte triées par PepMap : les étiquettes provenant d’une même protéine se regroupent au niveau du gène correspondant. Un véritable séparateur de protéines in silico…
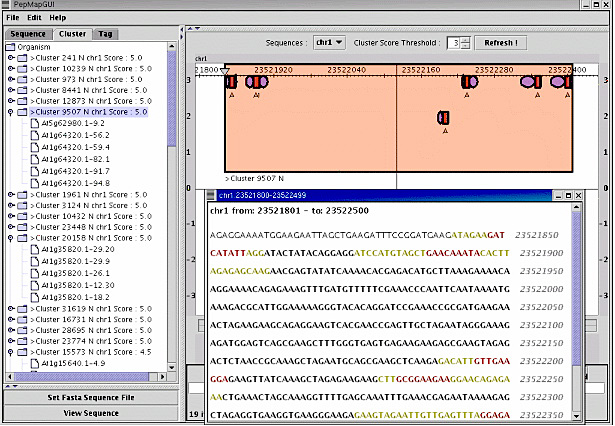
Exemple d’interface du logiciel PepMap.
Localisation d’un gène sur le génome d’« Arabidopsis thaliana ». Le panneau de gauche de la fenêtre principale de l’interface PepMap propose les meilleures localisation de l’étiquette peptidique. Le panneau de droite montre la localisation sur le génome de la sélection de la fenêtre de gauche. La fenêtre en option montre la séquence de nucléotides qui correspond à la localisation sélectionnée (les nucléotides en vert/rouge ne correspondent à aucune séquence en correspondance).

Plant d’Arabidopsis thaliana, ou Arabette des dames, écotype columbia.
© CNRS Photothèque – Arondel
Arabidopsis thaliana, ou Arabette des dames, est une plante de la famille des Brassicacées, à laquelle appartiennent de nombreuses espèces cultivées comme le chou, le navet, le radis, la moutarde, etc.
Côté génétique, A. thaliana a seulement cinq paires de chromosomes. La longueur de l’ADN d’une cellule est estimée entre 60 et 100 millions de paires de bases, c’est-à-dire environ cent fois moins que pour des plantes cultivées comme l’orge et le maïs. Plusieurs centaines de mutations sont connues, dont certaines sont visibles à l’œil nu (couleur, poils, fleurs etc.).
L’ensemble des caractéristiques de cette plante (culture facile, petite taille, croissance rapide, autopollinisation, descendants nombreux, petit génome, nombreuses mutations) ont conduit la communauté scientifique internationale à choisir cette plante comme l’un des organismes modèles utilisés en génétique. Un programme international de séquençage de son génome, auquel participent des laboratoires du monde entier, dont une trentaine en Europe, a été lancé en 1989.
Le développement de tels outils, puissants et rapides, est devenu indispensable. Ces logiciels sont capables de traiter avec fiabilité le flux d’informations engendré par les techniques biologiques d’analyse protéomique. Cependant, le « tout-automatique » n’est pas envisageable, le matériel vivant réserve des surprises qui nécessitent encore – heureusement – l’intervention de neurones non artificiels pour être appréciées à leur juste valeur !
Newsletter
Le responsable de ce traitement est Inria. En saisissant votre adresse mail, vous consentez à recevoir chaque mois une sélection d'articles et à ce que vos données soient collectées et stockées comme décrit dans notre politique de confidentialité
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

