Christian Gautier, un pionnier de la bio-informatique
Entretien avec Christian Gautier mené par Anne Lefèvre-Balleydier.

Vous avez opté pour la génomique à ses débuts. Par hasard ?
En partie. Comme je n’étais pas trop mauvais en mathématiques, on m’a encouragé dans cette voie. J’ai donc rejoint l’École normale supérieure en 1969, obtenu mon agrégation de mathématiques, puis un diplôme d’études approfondies (DEA) dans cette discipline. Mais j’avais en tête de faire de la biologie, et j’ai donc également passé un DEA de Génétique quantitative et appliquée. On m’a alors proposé un poste d’assistant à l’université de Lyon, en génétique. Là, j’ai fait ma thèse sous la direction de Richard Grantham, et j’ai commencé à travailler sur les problèmes d’interface entre mathématiques et génomique. Pourtant personnellement, j’étais au départ plus attiré par l’écologie. Finalement, la boucle est bouclée : je suis actuellement le président de la section d’écologie du CNRS…
Très vite, vous avez créé avec l’équipe de Richard Grantham la première banque de données sur les acides nucléiques, ACNUC. Quelle a été son histoire ?
Au début des années quatre-vingts, cette banque de séquences a d’abord été diffusée sous la forme d’un livre. Mais à peu près à la même époque, deux autres banques de données ont été créées, l’une aux États-Unis (GenBank, créée à Los Alamos et aujourd’hui sous la tutelle du National Center for Biotechnology Information ou NCBI), l’autre en Allemagne (créée à Cologne par K. Stüber). Or au niveau européen, il était logique de n’en maintenir qu’une, du fait des moyens humains alors requis pour la saisie des données. Et comme on ne pouvait pas obtenir ces moyens à Lyon, la base de données européenne s’est développée en Allemagne : ainsi est née la banque d’EMBL (Laboratoire européen de biologie moléculaire) qui s’est d’abord déplacée de Cologne à Heidelberg, avant de rejoindre l’Institut de bio-informatique européen (EBI), près de Cambridge.
ACNUC n’est pas morte pour autant. On l’a nourrie des données de GenBank et d’EMBL, tout en continuant de développer son système d’interrogation. Lyon est ainsi resté pendant très longtemps le seul lieu où l’on pouvait faire des requêtes complexes sur une base de séquences. À cette époque, Internet n’existait pas, et la diffusion était réalisée par implémentation locale, par exemple à l’institut Pasteur. Ceci étant, le maintien « en vie » d’ACNUC a été très bénéfique à notre équipe. Cela nous a d’abord procuré un avantage dans l’analyse globale des séquences, concrétisé par des résultats originaux concernant le mode d’évolution des génomes. Cela nous a ensuite fourni une base logicielle sur laquelle nous avons pu construire un environnement constamment renouvelé en fonction des thèmes scientifiques de l’équipe (phylogénie, analyse comparative des génomes, cartographie génomique…). ACNUC a sans aucun doute joué un rôle majeur dans l’orientation scientifique de l’équipe.
Toujours au sein de l’équipe de Richard Grantham, vous avez rapidement introduit en génomique une méthode bien connue des statistiques : l’analyse des correspondances…
Cela remonte à 1980. Et cette méthode a contribué à la naissance de la fameuse « hypothèse du génome ». En étudiant les séquences ARN issus de différents organismes, nous avons en effet montré par des analyses factorielles de correspondance que chaque génome a son propre style d’écriture. En clair, cela signifie que parmi un choix de codons synonymes (ceux qui codent un même acide aminé), un organisme préfère certains codons pour l’ensemble de ses gènes, on dit qu’il possède un « usage du code » qui lui est propre. Depuis la publication de ce résultat, de nombreux laboratoires ont analysé les usages du code de nombreux organismes, montrant la généralité du résultat mais aussi ses limites dans le cas d’organismes complexes, les mammifères par exemple.
Vous avez montré que la sélection naturelle agit précisément sur le style d’écriture…
C’était en 1982. On pensait alors que le choix des codons synonymes était sans importance, que la sélection naturelle opérait au niveau de la protéine codée. Mais de façon simultanée avec le Japonais Ikemura, nous avons alors mis en évidence avec Manolo Gouy le premier exemple d’une influence de la sélection naturelle sur le choix entre codons synonymes, c’est-à-dire d’une sélection naturelle ne portant pas sur la protéine, mais sur la manière dont elle était codée. Nos travaux portaient sur la bactérie Escherichia coli. Pour traduire un codon d’un ARN messager en acide aminé, il faut que celui-ci soit reconnu par un anticodon porté par un ARN de transfert (ARNt). Or nous avons remarqué que certains ARNt étaient fréquents, d’autres rares. Mais aussi que les gènes produisant beaucoup de protéines, donc souvent traduits, utilisaient de préférence des codons reconnus par les ARNt les plus fréquents. Et ce, quitte à modifier les protéines dont ils étaient responsables.
Votre équipe travaille également depuis assez longtemps sur le problème des isochores. Pouvez-vous nous en dire un peu plus ?
Les isochores sont d’assez longs fragments d’ADN de composition homogène en nucléotides qui séparent ou « segmentent » le génome en plusieurs parties : certaines sont par exemple riches en nucléotides A et T, d’autres comportent des taux plus ou moins élevés de nucléotides G et C. En 1988, sur la base d’une analyse statistique, nous avons révélé avec Dominique Mouchiroud et Giorgio Bernardi l’existence d’une forte variabilité de cette structure au sein des génomes de mammifères. Nous nous sommes donc interrogés sur l’origine de cette variabilité. Actuellement, sous l’impulsion en particulier de Dominique Mouchiroud et Laurent Duret dans notre équipe, c’est une hypothèse neutraliste qui semble la plus crédible : les isochores résulteraient de mécanismes mutationnels particuliers. Cependant, l’histoire des isochores est loin d’être encore complètement écrite, la simple recherche de leurs limites pose des problèmes difficiles. C’est actuellement le thème de recherche de Christelle Melo de Lima, une doctorante qui vient d’une formation initiale de mathématiques…
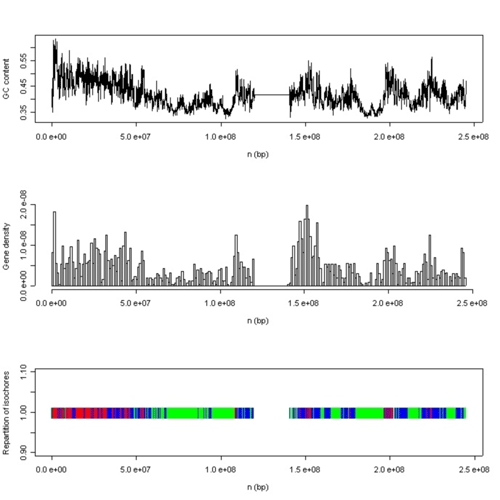
Structuration du chromosome 1 humain en isochores.
Le graphique du haut montre la variation du taux de G + C (pourcentage des lettres C et G) le long de la séquence du chromosome ; celui du milieu, la densité en gènes ; celui du bas, la répartition des isochores en trois classes : en rouge, les isochores de classe dite H (high) au sein desquels le taux de G + C est supérieur à 72%, en vert ceux de classe L (low, taux de G + C inférieur à 56%) et en bleu ceux de classe M (medium).
Source : Christelle Melo de Lima, Développement d’une approche markovienne pour l’analyse de l’organisation spatiale des génomes, thèse soutenue le 28 novembre 2005, université Claude Bernard de Lyon.
Vos recherches ont eu en partie pour moteur la théorie d’un grand généticien japonais…
On peut dire en effet qu’a contrario, la théorie neutraliste de Mooto Kimura a été un moteur. Notre équipe a cherché à tester cette théorie : c’est ce qui a déclenché nos recherches sur l’impact de la sélection naturelle sur le génome. M. Kimura a popularisé l’idée selon laquelle une grande partie des mutations se fixent par dérive génétique, sans procurer un avantage particulier aux individus qui les portent. Avant même les travaux de M. Kimura, un autre Japonais, Noboru Sueoka, avait proposé que les fréquences des bases dans les génomes reflétaient, en fait, des fréquences inégales des différentes mutations possibles (on parle de biais mutationnel). Nous avons donc cherché à voir dans quel cas la structuration du génome s’explique bien par la sélection naturelle : le style d’écriture d’E. coli en est un exemple. Mais dans beaucoup de cas, force a été de constater l’existence d’un biais mutationnel : un des exemples les plus clairs est la très faible fréquence avec laquelle un C (cytosine) est suivi d’un G (guanine) au sein du génome humain, structure qui résulte d’un fort taux de mutation d’un C quand il est suivi d’un G.
Aujourd’hui, les relations entre évolution et fonction, via l’analyse statistique de la structuration du génome, constituent toujours un de nos axes de recherche.
Quel autre axe de recherche développez-vous ?
Un axe important concerne le développement de modèles à l’interface entre biologie, mathématiques et informatique. Nous participons ainsi à un projet de cartographie comparée des génomes de vertébrés. On connaît en effet aujourd’hui les génomes de plusieurs vertébrés – Homme, souris, rat, chimpanzé, etc. Or leur comparaison s’avère très importante : quand on a établi la fonction d’un gène chez un organisme modèle, on peut éventuellement en déduire celle de son gène homologue chez l’Homme, avec de possibles retombées en médecine. Le problème, c’est qu’on ne peut pas comparer des génomes simplement en les accolant les uns aux autres. Car au cours de l’évolution, leur structure spatiale a été remodelée. De plus, pour une même espèce, il existe plusieurs systèmes de cartographie (cartes physique, génétique, cytologique mais aussi séquences…) qui ne sont pas toujours cohérents entre eux.
L’idée est donc de représenter toutes ces connaissances purement textuelles dans un cadre formel, de façon à pouvoir les implémenter sur une machine. Le but étant d’arriver à manipuler ensemble toutes les cartes, mais aussi les relations d’homologie connues entre différentes espèces, les données sur l’expression des gènes, sur leur polymorphisme… Un travail important de modélisation a été entrepris au sein du projet INRIA HELIX, en particulier en collaboration avec l’équipe de François Rechenmann à Grenoble. Une première base de données a été mise sur pied qui peut être consultée sur le Net depuis un an. Elle a d’ores et déjà permis d’obtenir des résultats concrets, bien qu’un peu hors du champ de recherches d’abord visé : en s’attachant à observer certains polymorphismes, Vincent Navratil et Abdel Aouacheria ont mis en évidence des polymorphismes spécifiques de cellules tumorales.
Le laboratoire que vous dirigez depuis 1997 a été créé au début des années soixante-dix par Jean-Marie Legay…
C’était un visionnaire, qui a créé une véritable école de biométrie lyonnaise. Avant lui, il y avait d’un côté des laboratoires de biométrie où travaillaient des mathématiciens, et de l’autre des biologistes qui collaboraient ponctuellement avec eux. Jean-Marie Legay a montré que l’interpénétration des deux disciplines était possible, et même fructueuse. Il a vu avant l’heure l’importance qu’il fallait accorder à la modélisation dans les systèmes biologiques. C’est donc quelqu’un qui a joué un grand rôle dans le devenir de tous les biométriciens lyonnais. Aujourd’hui, nous ne nous considérons ni comme des matheux, ni comme des biologistes. Nous avons les deux cultures, tout en ayant des connaissances en informatique, car elle intervient à la fois comme puissance de calcul et langage de modélisation. Naturellement, il y a au sein du laboratoire une sorte de gradient de compétences entre ceux qui ont plus l’habitude de la paillasse, et viennent plutôt de la biologie, et ceux qui sont plus dans la théorie, qu’elle soit mathématique ou informatique. Mais c’est grâce à ce gradient que des interactions se produisent !
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !
