

sous licence Creative Commons
Indexer les données génomiques : un défi de taille à relever
Les ensembles de données génomiques publiques issus de la lecture des génomes augmentent à un rythme exponentiel. Ils contiennent des trésors d’informations qui permettent des découvertes révolutionnaires dans des domaines fondamentaux tels que l’agronomie, l’écologie et la santé. Malheureusement, malgré leur disponibilité publique dans des banques telles que le « European Nucleotide Archive » (ENA), ces ressources, mesurées en pétaoctets, sont rarement réutilisées au niveau mondial parce qu’elles ne peuvent pas faire l’objet d’une requête. Regardons de plus près une solution permettant de faire des requêtes sur les données générées par le projet Tara Oceans, qui représentent déjà à elles seules plusieurs dizaines de téraoctets de données.
L’ADN : ses données génomiques et métagénomiques
Les cellules des organismes vivants portent de l’information génétique sous forme d’ADN. Cette information est utilisée par le mécanisme cellulaire pour, entre autres, produire des protéines et réguler l’expression des gènes, et, au final, déterminer le phénotype de l’organisme. Par phénotype on entend des observations allant des caractéristiques biochimiques d’un organisme jusqu’à l’ensemble des traits apparents d’un individu (taille, couleur, résistance à certaines maladies…).
Être en mesure de lire l’ADN est crucial pour comprendre finement le fonctionnement du vivant. On peut par exemple mentionner la pandémie de Covid-19, où l’accès à l’ADN a permis de comprendre et de limiter l’évolution de la maladie, et de mettre rapidement en œuvre des solutions pharmaceutiques. Un dossier Interstices est spécifiquement dédié à ce sujet.
La lecture de l’ADN à partir d’échantillons biologiques est rendue possible grâce aux séquenceurs. Ces machines produisent un texte, sur l’alphabet de l’ADN (A,C,G,T) qui représente l’information génétique contenue dans les cellules de l’échantillon séquencé. Les séquenceurs ne produisent que de courtes séquences de quelques centaines de caractères chacune, appelées des lectures. Pour chaque lecture, son information de position originale sur le génome est inconnue. De plus, ces séquences sont partiellement erronées. La reconstruction des génomes à partir des données de séquençage brutes consiste à retrouver l’ordre des lectures. Cette opération de reconstruction, appelée l’assemblage, est décrite dans cet article Interstices.
Un génome assemblé est représentatif d’une espèce. Pour autant, il est loin de représenter la totalité des informations présentes dans les données brutes. En effet, l’assemblage des lectures fournit un consensus où les petites variations entre cellules ou chromosomes sont perdues. Plus généralement, une large partie des lectures originales ne sont pas utilisées pour l’assemblage.
Cette situation est d’autant plus vraie lorsque l’échantillon séquencé ne provient pas d’un unique individu ou d’une unique espèce, mais qu’il provient d’un milieu complet (un écosystème) tel qu’un verre d’eau de mer par exemple. C’est ce que l’on nomme la métagénomique. Dans cette situation, on peut mélanger plusieurs millions de cellules lors du séquençage. On ne sait alors pas à quelle espèce appartient chaque lecture fournie par le séquenceur. Dans le cadre de la métagénomique, étant donnée la complexité des données, il est généralement impossible de reconstruire les génomes des espèces présentes.
Pour toutes ces raisons, que ce soit en génomique ou en métagénomique, les données brutes de séquençage sont conservées et la majorité est publiquement disponible. Par exemple le service européen « European Nucleotide Archive » (ENA) contient en Juillet 2023 plus de 65 Pétaoctets de telles données brutes publiques, soit environ l’équivalent de cent mille disques durs d’ordinateurs de bureau classiques.
Bien que ces données soient publiquement disponibles, il n’existe malheureusement pas de moteur de recherche pour y faire des requêtes. Si vous cherchez à découvrir si une séquence qui vous intéresse, disons “ATGAGAAAAGTAGCAATTTACGGAAAAGGC”, existe quelque part, et où, il est impossible de répondre à cette question. Finalement nous pouvons imaginer la situation de ce trésor d’informations contenues dans les banques de données génomiques comme ce que serait Internet sans pouvoir utiliser de moteur de recherche : largement sous-exploité.
Indexer pour rechercher
Lorsque l’on cherche un mot sur Internet, le moteur de recherche ne scanne pas l’intégralité des pages web pour y détecter dans chacune d’entre elles si le mot recherché y apparaît. Il utilise un index, c’est-à-dire une structure de données qui associe chaque mot à un ensemble de sources dans lesquelles il apparaît. L’index permet de connaître les localisations d’un mot dans un gros ensemble de données en un temps qui ne dépend pas de la taille des données indexées. Idéalement, il fournit en quelques millisecondes la liste des documents dans lesquels apparaît un mot recherché. Ainsi lorsque l’on cherche une phrase via un moteur de recherche sur le Web, l’index fournit pour chaque mot de la phrase une liste des pages dans lesquelles il apparaît. Le moteur de recherche combine ensuite ces résultats pour offrir une réponse à l’utilisateur/utilisatrice.
Précisons la terminologie : dans la suite, un ensemble de données est composé d’entités qui seront appelées des « documents ». Dans l’exemple du moteur de recherche, l’ensemble des données est composé des pages web qui sont chacune un document. Dans le cas de la génomique évoquée par la suite, un document peut être un génome ou un jeu de données brutes généré par le séquençage d’un individu ou d’un environnement. On dira que l’on interroge l’index avec un mot pour trouver les documents dans lesquels ce mot apparaît.
L’objectif présenté ici est de proposer un index permettant de créer un moteur de recherche efficace pour des milliers de documents, où chacun est le résultat brut du séquençage d’un métagénome, composé de centaines de millions de lectures.
Un moteur de recherche pour les données brutes génomiques : que sait-on faire et pourquoi est-ce complexe ?
Il existe déjà un moteur de recherche (BLAST) capable de faire des requêtes sur les données de génomes assemblés. Au prix de quelques minutes d’attente, il est par exemple possible de savoir que notre séquence d’intérêt “ATGAGAAAAGTAGCAATTTACGGAAAAGGC” fait partie notamment de la bactérie Desulfovibrio. Vous pouvez essayer, tout est prêt pour vous, cliquez simplement sur le bouton “BLAST” en bas de la page.
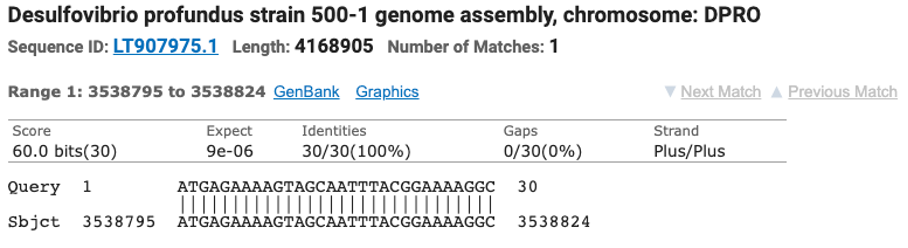
Exemple de résultat fourni par le serveur Blast au NCBI.
Ce service est extrêmement précieux mais il se limite aux génomes assemblés, dont la taille cumulée est environ cinq cent mille fois plus petite que les 65 pétaoctets des données disponibles dans l’ENA. Utiliser cette approche pour faire des recherches dans ces données brutes prendrait plusieurs centaines de jours, et aucun service ne le propose.
On peut se demander pourquoi ne pas simplement adapter aux données génomiques les moteurs de recherche conçus pour le Web ? La réponse est simple. Les données génomiques sont partiellement erronées, et très redondantes. Mais surtout elles ne contiennent pas de mot. À l’inverse, le langage naturel est pratique à indexer. La langue française par exemple utilise quelques milliers de mots. On sait assez facilement associer chaque mot à une liste de pages web où il apparaît. Ceci s’implémente par exemple par l’utilisation d’une table de hachage.
Pour créer une notion de mot dans les données génomiques ou métagénomiques, nous définissons arbitrairement un concept de mot de taille fixe : les k-mers.
Les k-mers et leur utilisation
Un 𝑘-mer désigne simplement “un mot de taille 𝑘”. Par exemple, si on prend 𝑘=3, la séquence “ATTGC” est composée des 3-mers “ATT”, “TTG”, et “TGC”. On utilise en général une valeur de k suffisamment grande pour que les k-mers d’une séquence ou d’un document leur soient spécifiques. En pratique la valeur utilisée est autour de 𝑘 = 30.
Les 𝑘-mers sont partout lorsque l’on propose des algorithmes de traitement de données génomiques. Dans notre situation, le principe est simple. Le nombre de 𝑘-mers partagés entre une séquence requête et un document donne une approximation de leur similarité. Cette notion de similarité approchée par les 𝑘-mers est décrite largement dans l’article Analyser les génomes des océans.
Donc pour mettre en œuvre un moteur de recherche, l’opération fondamentale que l’on doit être en mesure d’effectuer efficacement est “Dans quel(s) document(s) ce 𝑘-mer existe t’il ?”.

Exemple d’utilisation d’un index. Ici l’index est interrogé avec le k-mer ACTTAGA. L’index indique que ce 𝑘-mer existe dans les documents 𝑏8,𝑏42,…𝑏8728.
Formellement, on note 𝑞 une requête, et 𝐵 = {𝑏1,𝑏2,…,𝑏𝑛} la banque composée de 𝑛 documents (nommés de 𝑏1 à 𝑏𝑛). Lors de la requête, l’index est interrogé avec chaque k-mer de 𝑞. Ainsi chaque k-mer est associé à l’ensemble des documents dans lesquels il apparaît (par exemple 𝑏8,𝑏42,…𝑏8728 dans la figure précédente). Au final, on connaît pour chaque document 𝑏𝑖 le nombre de k-mers qu’il a en commun avec 𝑞, ce qui permet de faire une approximation de sa similarité avec 𝑞.
On peut alors se poser de nouveau la question : pourquoi ne pas utiliser les moteurs de recherche issus du Web pour indexer ces k-mers ? La réponse est liée à la quantité de k-mers. L’alphabet des séquences génomiques comporte quatre lettres (A,C,G,T). Il y a donc 4𝑘 k-mers différents. Avec k = 30, il y a environ 1018 k-mers différents, soit plusieurs centaines de millions de milliards. On est très loin de la situation des langages naturels avec leurs quelques dizaines de milliers de mots, et il est impensable d’en utiliser les outils.
Les données de séquençage d’un échantillon peuvent contenir plusieurs centaines de milliards de k-mers distincts. L’index pourrait être composé d’une table de hachage permettant d’associer un k-mer aux documents dans lesquels il apparaît. Mais les tables de hachage sont trop gourmandes en espace pour passer à l’échelle sur les données génomiques. On a donc recours à une structure de données dite probabiliste — les filtres de Bloom, ainsi qu’une astuce pour réduire leur taux de faux positif (voir l’article Indexer des milliards d’éléments avec les filtres de Bloom).
En pratique les k-mers d’un document sont indexés grâce à un filtre de Bloom. Ainsi, si l’on souhaite indexer 𝑛 documents, on crée 𝑛 filtres de Bloom distincts.
Organiser l’index pour qu’il soit efficace
Avec les filtres de Bloom, nous avons une solution pour indexer efficacement les k-mers d’un document. Il s’agit maintenant d’étendre cette structure à un ensemble de documents pour permettre l’indexation de grands ensembles de données. Pour cela, on peut imaginer une matrice de filtres de Bloom où chaque ligne est un filtre correspondant à un document. Ce type de structure permet de construire facilement l’index par simple concaténation des filtres correspondant à chacun des documents.
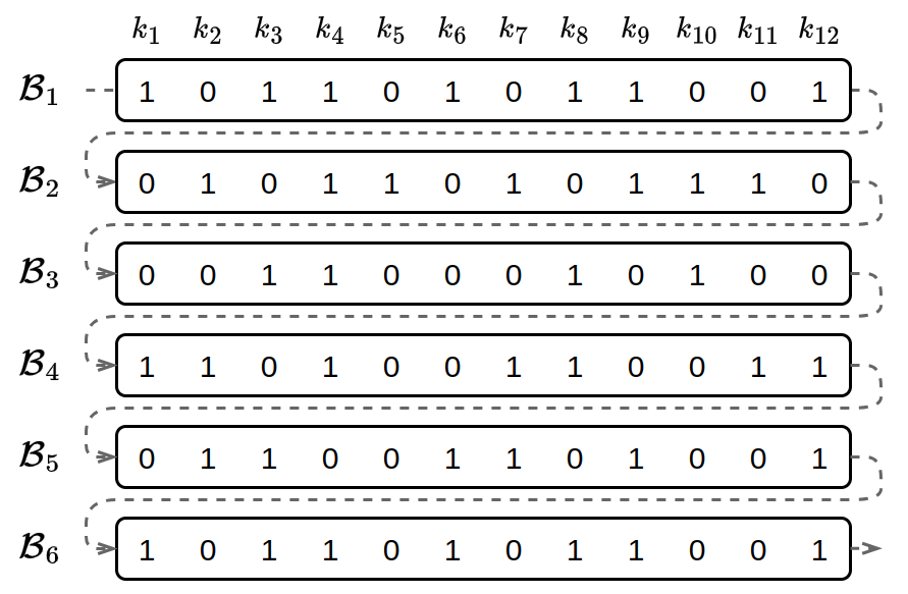
Représentation simplifiée d’un ensemble de filtres de Bloom (𝐵1…𝐵6) pour indexer 6 documents avec 12 k-mers (k1,…,k12). L’index « simplifié » est un tableau de bits, le bit 0 ou 1 d’une case du tableau indique, avec un taux de faux-positif non nul si un k-mer est contenu dans le document.
Malheureusement, cette organisation n’offre pas les meilleures performances. La recherche d’un k-mer dans un document consiste uniquement à vérifier la valeur d’un bit à une position donnée de chaque filtre. Cette opération est triviale mais demande le transfert de ce bit depuis le support stockage de l’index vers le processeur qui vérifiera sa valeur. En conséquence, c’est la vitesse à laquelle nous sommes capables de lire ces bits qui va déterminer les performances de notre algorithme. Ces transferts entre unités de calcul et supports de stockage sont appelés « opérations d’entrées/sorties ». Lorsqu’un algorithme est limité par la vitesse de ces opérations, les importants moyens de calcul dont nous disposons aujourd’hui, toujours plus nombreux et puissants, ne sont pas d’une grande aide.
Pour améliorer la vitesse d’accès aux données, les architectures modernes utilisent le concept de cache. Le cache est un espace de stockage temporaire permettant de conserver une copie des données provenant d’une source moins performante afin de diminuer le coût d’éventuels accès ultérieurs. Un ordinateur dispose de trois espaces de stockage principaux : le disque dur, la mémoire vive et la mémoire du processeur. Pour réaliser un calcul sur des données, celles-ci doivent transiter par ces différents espaces pour rejoindre l’unité de calcul, chaque niveau servant de cache pour le niveau inférieur. Le transfert de données vers un niveau supérieur ayant un coût non négligeable, on charge souvent un peu plus de données que nécessaire afin de les conserver en cache. Pour profiter de ce système, il est donc important de stocker consécutivement les données qui ont une chance d’être demandées de manière successive. Ainsi, on minimise le nombre d’accès au support de stockage. Avec une organisation des filtres en lignes, les bits d’un k-mer spécifique correspondent à une colonne et sont donc séparés par un nombre de bits correspondant à la taille des filtres comme c’est le cas des 2 bits entourés dans la figure suivante. Sachant que chaque filtre de Bloom est constitué de plusieurs milliards de bits, on comprend que chaque accès à l’index n’apportera qu’un seul bit d’information.
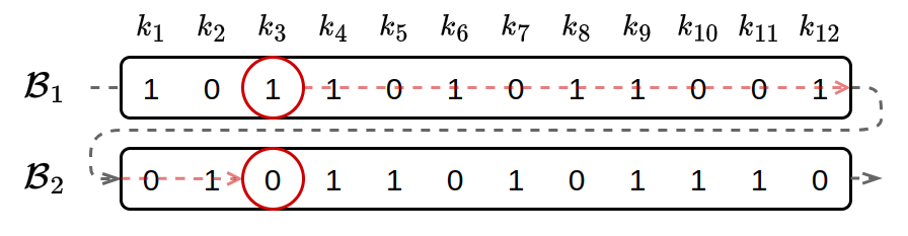
Avec une représentation des filtres de Bloom en lignes, les bits observés lors de l’interrogation d’un k-mer sont distants dans l’index.
En modifiant la structure de l’index, nous pouvons utiliser le cache à notre avantage. Du point de vue de l’organisation en mémoire, il s’agit de stocker de manière continue tous les bits correspondant à un k-mer spécifique, comme présenté dans l’exemple ci-dessous.
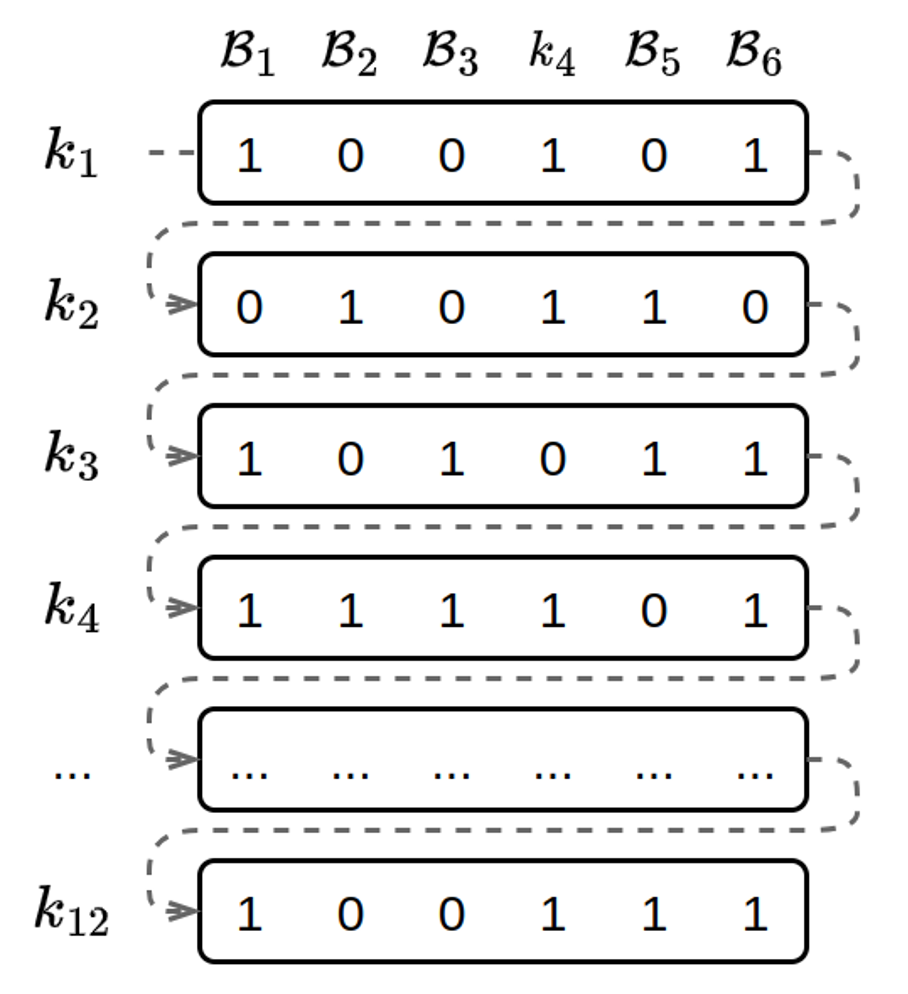
Avec une représentation des filtres de Bloom en colonnes, les bits observés lors de l’interrogation d’un k-mer sont consécutifs dans l’index.
Cette représentation correspond à la transposition de la matrice que nous avons vue précédemment. Ici chaque ligne représente la présence ou l’absence d’un k-mer dans chacun des documents. On appelle cette structure un index inversé.
Interroger l’index avec un k-mer consiste alors en la lecture de 𝑛 bits consécutifs à une position donnée, où 𝑛 est le nombre de documents dans l’index. Cette disposition offre de bien meilleures performances lors de l’interrogation de l’index par un k-mer unique.
Cependant le problème de dispersion de l’information n’est pas encore résolu. En effet, les séquences biologiques sont constituées de nombreux k-mers qui seront interrogés de manière successive. Or ces k-mers successifs peuvent se trouver n’importe où dans l’index.
Pour résoudre ce problème, l’idée est d’indexer les k-mers qui se chevauchent dans une zone spécifique de l’index. Pour ce faire, on utilise le concept de “minimizer”. Le minimizer d’un k-mer est le sous-mot de taille m, avec 𝑚 < 𝑘, qui minimise une fonction donnée, par exemple le plus petit mot au sens alphabétique. Une propriété intéressante est que deux k-mers successifs partagent la majorité de leurs m-mers, à l’exception du premier m-mer du premier k-mer et du dernier m-mer du second k-mer. Autrement dit, la probabilité pour que des k-mers chevauchants partagent un même minimizer est élevée. En choisissant une fonction de hachage adaptée, il est possible d’indexer les k-mers partageant un minimizer dans une même zone de l’index. On obtient alors un index constitué de plusieurs partitions correspondant à des sous-ensembles de minimizers spécifiques. Lors de la recherche d’une séquence, cela a pour effet de concentrer les k-mers interrogés dans des zones spécifiques. Au fur et à mesure de l’interrogation de l’index, ces zones vont progressivement monter dans le cache. Les interrogations ultérieures seront alors plus efficaces car elles demanderont peu ou pas d’accès au support de stockage, les réponses étant déjà présentes dans le cache. En plus d’améliorer les performances, cette technique permet aussi de paralléliser simplement les calculs au moment de la construction de l’index, chaque partition pouvant être construite de manière indépendante.

Deux représentations possibles des filtres de Bloom, à gauche en lignes, et à droite, en colonnes.
Les deux représentations possibles sont présentées sur la figure ci-dessus avec, à gauche, les filtres de Bloom en lignes, et à droite, les filtres de Bloom en colonnes. À titre d’exemple, supposons que nous sommes capables d’obtenir 4 bits d’information par lecture, on remarque que l’interrogation du k-mer 𝑘3 nécessitera six accès au support stockage dans le premier cas, et seulement deux dans le second. L’index inversé permet donc de réduire les accès au stockage qui représentent la majorité du coût d’une interrogation.
Un nouveau serveur public : Ocean Read Atlas (ORA)
Ces avancées ont permis d’indexer les k-mers des données métagénomiques issues du projet Tara Oceans et de proposer un serveur public, Ocean Read Atlas, permettant d’y faire une requête. En un clic, plusieurs dizaines de téraoctets de données (originalement stockées sur ENA) peuvent être interrogées. On peut donc connaitre la répartition d’une séquence d’ADN à travers tous les océans du globe. Auparavant, effectuer une telle recherche nécessitait plusieurs semaines de calculs sur des supercalculateurs.
L’utilisateur indique une requête composée d’une ou plusieurs séquences d’ADN. En réponse, le serveur indique parmi les 1393 échantillons d’eau de mer lesquels sont susceptibles de contenir une ou plusieurs séquences de la requête.
Ce site web permet un temps de réponse de l’ordre de la seconde pour une requête de 1000 séquences. En plus des résultats numériques, les réponses sont présentées sous forme de cartes ou graphiques interactifs représentant la présence de séquences similaires dans les stations de prélèvement en fonction de leurs propriétés environnementales (température, salinité, oxygène, etc.).
Par exemple, la carte, ci-dessous, représente la répartition biogéographique des séquences qui partagent les k-mers contenus dans la séquence du gène appelé nifH de l’espèce Pseudodesulfovibrio profundus.
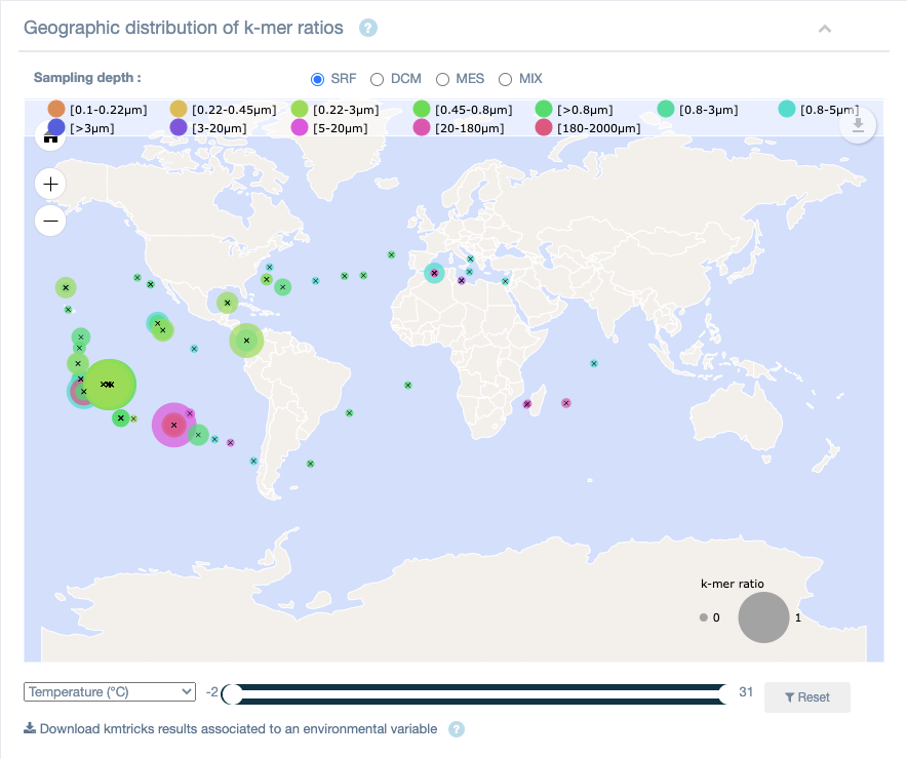
Résultat d’une recherche sur Ocean Read Atlas : répartition biogéographique des séquences recherchées.
Les résultats de ce type de requêtes peuvent également être représentés en fonction de la colonne d’eau (de la profondeur où ont été prélevés les échantillons : SRF pour Surface, DCM pour le Deep Chlorophyll Maximum (Maximum profond de Chlorophylle), ou MES pour la zone mésopélagique). En faisant varier les variables environnementales (ci-dessous la température) et leurs valeurs, on peut identifier si le gène ou le génome que l’on recherche se trouve dans un environnement particulier. Dans le cas du gène recherché ici, on le trouve plutôt dans des eaux de surface et chaudes (entre 25 et 30 °C).
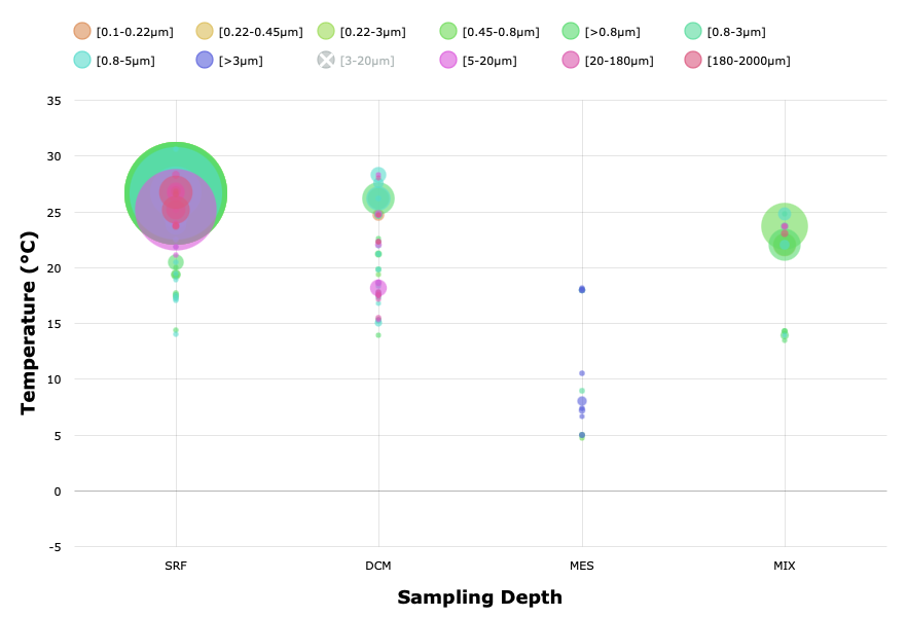
Résultat d’une recherche sur Ocean Read Atlas : répartition de la température de l’eau où la séquence recherchée apparaît.
La suite
Les avancées présentées ici ont permis de changer d’ordre de grandeur, en indexant des dizaines de téraoctets de données brutes. Les bases de données publiques en contiennent actuellement des pétaoctets (mille fois plus). Il faut donc poursuivre les efforts de recherche notamment en termes de distribution de calculs, et de découverte de nouvelles structures de données.
Aujourd’hui nous savons indiquer si un k-mer existe ou non dans un document. Mais nous ne savons pas lui associer d’information telle que “dans quelles lectures apparaît-il ?”, “existe-t’il de l’information associée à sa séquence ?”… À l’avenir, il conviendra de trouver des méthodes permettant de telles associations tout en limitant l’impact mémoire de cette fonctionnalité.
Ces travaux ont été financés par le projet ANR SeqDigger (ANR-19-CE45-0008) ainsi que par le Défi Inria OmicFinder.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !


Magali Lescot

Pierre Peterlongo
Directeur de recherche Inria, responsable de l'équipe de recherche GenScale au centre Inria de l'Université de Rennes.