

sous licence Creative Commons
Comment la bioinformatique a résolu le puzzle du génome du SARS-CoV-2
Séquençage du matériel génétique
L’identification de l’agent causal d’une infection inconnue passe généralement par le séquençage de son matériel génétique, c’est-à-dire la détermination de la séquence des nucléotides (les A, T, C et G) qui composent son génome. Depuis le milieu des années 2010, le séquençage à haut débit couplé à la bio-informatique permet de caractériser le génome d’un virus émergent en quelques jours pour quelques centaines d’euros. Ainsi, de décembre 2019 à janvier 2020, plusieurs hôpitaux de Wuhan en Chine confrontés à la maladie se sont lancés indépendamment dans le séquençage du nouvel agent pathogène, en suivant approximativement le même protocole. Le point de départ consiste à collecter le fluide pulmonaire de patients, puis à en extraire le matériel génétique. L’ADN ainsi obtenu est prêt à être séquencé. Lors du séquençage, les molécules d’ADN ou d’ARN ne peuvent pas être obtenues de bout en bout car la technologie utilisée ne produit que de courts fragments de séquences d’environ 200 nucléotides chacun. À l’issue du séquençage, les données brutes sont ainsi une soupe de centaines de millions de courtes séquences nucléiques, appelées lectures, qui couvrent de manière aléatoire les génomes initiaux. Reconstituer le génome d’intérêt nécessite ensuite toute une série de traitements informatiques.
Identification des lectures d’intérêt
Le premier problème est que les données de séquençage sont mélangées. Elles proviennent de tous les micro-organismes présents dans l’échantillon clinique possiblement associés aux cellules humaines environnantes. Il faut donc faire le tri dans ce microbiome pulmonaire. Cela est réalisé par la fouille de grandes bases de données génomiques comprenant les génomes de l’ensemble des microbes connus (virus, bactéries, champignons et parasites) ainsi que le génome humain. Pour cela, la communauté bio-informatique a développé dès la fin des années 1990 des moteurs de recherche génomique, tels que Blast, capables de traiter efficacement des grandes masses de séquences à l’image d’un Google pour ADN. Ces outils calculent des alignements de séquences qui identifient les lectures similaires aux séquences présentes dans la base de données et distinguent ainsi les lectures provenant potentiellement du nouveau virus (voir la figure 1 ci-dessous, à droite). Les méthodes les plus récentes ont été conçues depuis 2009-2010 pour traiter spécifiquement les lectures de séquençage à haut débit. Le cœur algorithmique s’appuie sur des concepts avancés issus de la théorie de l’information, tels que la compression, les fonctions de hachage, les structures de données d’indexation. On peut ainsi en quelques minutes de calcul isoler la fraction des lectures provenant du nouveau virus, fraction qui représente généralement moins de 1 % des données initiales.
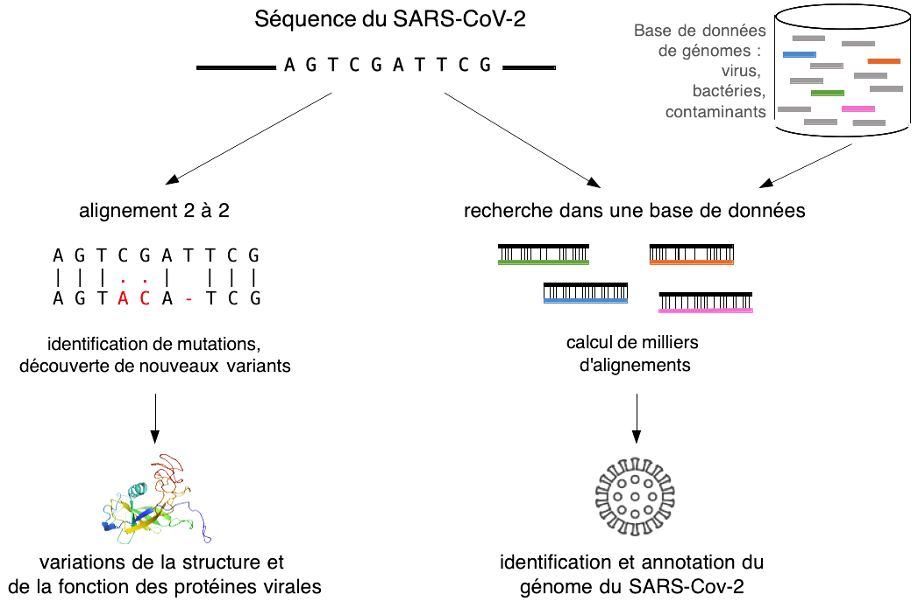
Figure 1 : Alignement de séquences biologiques.
Un problème fondamental en bio-informatique : l'alignement de séquences d'ADN
Est-ce que des séquences d’ADN se ressemblent ? De « combien » se ressemblent-elles ? C’est en alignant les séquences qu’on peut répondre à ces questions (voir figure 1 ci-dessus, à gauche). L'alignement est le processus algorithmique qui consiste à comparer des séquences, nucléotide par nucléotide, pour identifier les similitudes et les différences. Dans le cas de séquences similaires, les différences correspondent à des mutations, qui sont des événements évolutifs naturels, ou à des erreurs de séquençage : remplacement d'un nucléotide par un autre, insertion d'un nucléotide supplémentaire ou suppression d'un nucléotide.
Comment chercher une aiguille dans une botte de foin ?
Pour identifier l’alignement optimal, qui minimise le nombre de différences entre deux séquences, il est nécessaire d’explorer toutes les combinaisons d’insertions ou suppressions possibles. Néanmoins il en existe un nombre exponentiel en la longueur de la séquence. Par conséquent, l'approche naïve consistant à calculer tous les alignements est infaisable en pratique. L’alignement est résolu comme un problème d'optimisation en utilisant la programmation dynamique, un paradigme algorithmique classique. L’idée est de diviser le problème en sous-problèmes plus petits et de calculer ainsi l'alignement optimal sans recourir à des approximations.
Des alignements peuvent également être calculés entre une séquence requête et une base de données contenant des millions de génomes (voir figure 1 ci-dessus, à droite). Cela permet par exemple d’identifier dans la soupe initiale des lectures de séquençage d'un échantillon pulmonaire celles provenant du génome humain et d’autres parasites respiratoires connus, afin de les écarter. Pour de telles comparaisons à grande échelle, le calcul des alignements exacts par programmation dynamique nécessiterait des mois de calcul. Pour surmonter cette difficulté, les chercheurs et chercheuses en bio-informatique ont proposé des heuristiques efficaces capables de traiter des gigaoctets de données ADN avec un ordinateur de bureau. Le calcul est effectué en organisant la base de données en une structure d'indexation qui répertorie tous les mots d'une longueur donnée, appelés k-mers, provenant des génomes à indexer et qui permet un accès direct à ces mots, à la manière d’un dictionnaire. La conception de telles structures d’index est un sujet de recherche actif. À titre d’exemple, on peut citer les approches par hachage.
Après avoir rapidement identifié les séquences qui partagent des k-mers, des alignements plus précis et plus longs peuvent être réalisés par programmation dynamique. Ce type d'approche permet d'identifier une aiguille dans une botte de foin : des similarités aussi courtes que dix nucléotides entre la séquence d'intérêt et la base de données.
Signification statistique des alignements
Mais, avec de grands volumes de données, des correspondances peuvent se produire par hasard, sans signification biologique. Il est donc crucial d'évaluer la signification statistique des alignements trouvés, ce qui est fait avec des calculs de E-valeurs qui mesurent le nombre d'alignements attendus du seul fait du hasard. Plus ces valeurs sont proches de 0, et moins la correspondance est imputable au hasard. Par exemple, il est possible de trouver des similitudes locales entre le gène de la protéine Spike du SARS-CoV-2 et le génome d’organismes de l'arbre du vivant aussi divers que les bactéries Escherichia coli et Bacillus subtilis, le maïs, le poisson zèbre, ou même un virus non apparenté, comme le VIH. Ces correspondances ont toutes une E-valeur supérieure à 0,01, ce qui n'est pas significatif, tandis que l'alignement entre le gène spike du SARS-CoV-2 et d'autres gènes spike de coronavirus atteint une E-valeur des milliards de milliards de fois plus faible.
Assemblage du génome
Une fois que les lectures d’intérêt ont été isolées, la seconde étape est l’assemblage, c’est-à-dire la reconstruction de la séquence du génome à partir du puzzle des lectures. L’assemblage d’un nouveau génome s’apparente au montage d’un meuble en kit dont le mode d’emploi aurait été perdu : il y a toutes les pièces, mais pas les instructions. La reconstruction est encore compliquée par le fait qu’il y a des centaines de milliers de pièces qui se ressemblent, car écrites sur le même alphabet à quatre lettres, A, C, G, T, et que certaines contiennent des erreurs dues au séquençage. L’assemblage de novo reste un défi majeur pour les grands génomes de plusieurs milliards de nucléotides. Mais les logiciels développés au cours de la dernière décennie peuvent désormais résoudre facilement le puzzle de l’assemblage pour les petits génomes, tels que les génomes viraux. Les méthodes actuelles reposent sur les graphes de De Bruijn, dont nous donnons une brève introduction dans la figure 2 ci-dessous. L’assemblage aboutit à la reconstruction de la séquence génomique du virus : dans le cas du SARS-CoV-2, il s’agit d’une séquence ARN simple brin composée d’environ 30 000 nucléotides. La première séquence de référence a été rendue publique le 12 janvier 2020. Au total, il aura fallu moins de deux semaines pour obtenir le génome de ce que l’on appelle désormais le virus SARS-CoV-2.
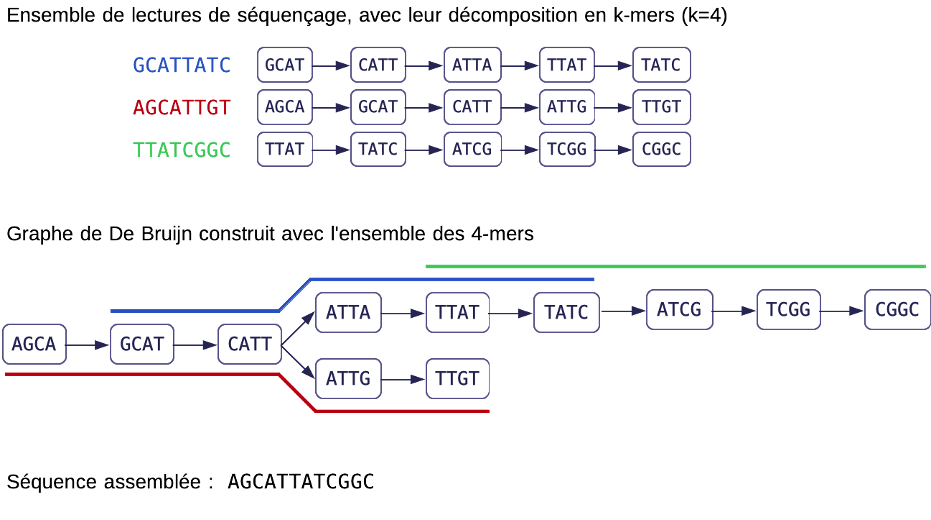
Figure 2 : Le graphe de De Bruijn, pour résoudre le puzzle des lectures de séquençage.
Le principe du graphe de De Bruijn appliqué au problème de l'assemblage
La méthode la plus couramment employée pour reconstruire un génome à partir de lectures de séquençage repose sur un « graphe de De Bruijn ». Ce nom vient du mathématicien Nicolaas de Bruijn, qui a introduit cette structure de données dans les années 1940 en tant qu'objet combinatoire. Le graphe de De Bruijn a fait son entrée en bio-informatique soixante ans plus tard, avec l'avènement du séquençage à haut débit. En effet, la quantité de données de séquençage générées classiquement par une expérience (gigaoctets, voire téraoctets) a rendu les méthodes obsolètes et a nécessité un nouveau paradigme pour l'assemblage des génomes.
Les lectures sont découpées en mots d'une longueur fixe k, les k-mers, qui sont plus courts que les lectures entières. Dans l'exemple ci-dessus, nous avons pris k = 4, mais en pratique, la valeur de k varie entre 20 et 130 de telle sorte que la majorité des k-mers soient présents une seule fois dans le génome à reconstruire. Le graphe de l'ensemble des lectures est alors construit en prenant chaque k-mer unique comme sommet et en ajoutant une arête entre deux sommets si les k-mers se chevauchent exactement de k−1 nucléotides. Ce faisant, la taille du graphe ne dépend que du nombre de k-mers distincts, plutôt que du nombre total de lectures. Cela fait une économie conséquente. De plus, les arêtes peuvent être très rapidement calculées et ne nécessitent pas d'être stockées explicitement puisqu'elles sont déduites de la liste des sommets.
La séquence originale du génome est obtenue comme un chemin dans ce graphe. Ce parcours de graphe est lié à la recherche de chemins eulériens, qui sont des chemins où chaque arête est visitée exactement une fois, tout en permettant de visiter plusieurs fois un sommet si nécessaire. Calculer le chemin eulérien est un problème facile, et il existe un algorithme en temps proportionnel au nombre de sommets du graphe pour le calculer. La méthode doit cependant être adaptée pour prendre en compte le caractère expérimental des données : erreurs de séquençage, régions génomiques répétées ou non couvertes par le séquençage.
Et ensuite ?
Rétrospectivement, la détermination de la séquence du génome du SARS-CoV-2 pourrait presque sembler anodine tant la tâche a été réalisée rapidement et sans difficulté particulière. Pourtant une vingtaine d’années auparavant, le travail aurait été autrement plus ardu. Il aurait probablement fallu des mois pour y parvenir. Entre-temps, les technologies de séquençage de l’ADN se sont améliorées et démocratisées, des outils bio-informatiques pour le traitement de ces données ont été développés et diffusés sous licence libre, les rendant accessibles à tout le monde.
La détermination de la séquence du génome est une première étape obligatoire, mais qui en elle-même n’est que peu informative. Il faut ensuite donner du sens au génome obtenu pour comprendre son organisation et le fonctionnement du virus. Ce sont des étapes essentielles pour pouvoir surveiller l’évolution des gènes clés du virus et l’apparition de variants. La connaissance de la séquence du génome permet également le développement de tests de diagnostic, indispensables au contrôle de l’épidémie.
- Le premier génome de référence publié est disponible sur le site du NCBI avec l’identifiant NC_045512.
- SARS-CoV-2 et Covid-19 : jouons sur les mots, Camille Marchet, blog Binaire, 6 mai 2020.
- Alignement optimal et comparaison de séquences génomiques et protéiques, François Rechenmann, Interstices, 2005.
- Le hachage, Christian Schindelhauer, Interstices 2009.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !



Claire Lemaitre
Chargée de recherche Inria en bioinformatique au sein de l'équipe Genscale au centre Inria de Rennes.