

sous licence Creative Commons
Décoder le génome : vers la compréhension du fonctionnement du SARS-CoV-2
Déterminer la séquence du génome du virus, présentée dans l’article Comment la bio-informatique a résolu le puzzle du génome du SARS-CoV-2, a été une étape majeure dans l’étude de la maladie. En premier lieu, cela ouvre la voie à l’analyse phylogénétique du virus, qui identifie les virus proches partageant des relations évolutives. En effet, la comparaison des séquences à grande échelle avec les bases de données virales a révélé peu après son séquençage que le génome de ce mystérieux virus est un nouveau bétacoronavirus, l’un des quatre genres de coronavirus. Il présente plus de 85 % de similarité avec plusieurs coronavirus dérivés de chauves-souris, tout en étant plus éloigné des autres bétacoronavirus humains connus. Il présente ainsi 79 % de similarité avec le SARS-CoV, responsable de l’épidémie de SRAS en Asie en 2003, et 50 % de similarité avec le coronavirus du syndrome respiratoire du Moyen-Orient, le MERS-CoV. La disponibilité de tous ces génomes de virus apparentés permet de formuler des hypothèses préliminaires sur l’origine du virus. Elle permet également de caractériser les protéines codées par le génome qui régissent le fonctionnement du virus et donc d’envisager des moyens de lutter contre la maladie.
Un court rappel sur le cycle viral
Un virus ne peut pas fonctionner de manière autonome, par lui-même. Sa survie dépend des cellules hôtes. Pour cela, il détourne la machinerie cellulaire de l’hôte pour se multiplier et infecter d’autres cellules. Ce cycle de vie repose généralement sur cinq étapes :
- la reconnaissance par le virus des protéines réceptrices à la surface des cellules de l’hôte, puis l’attachement à ces protéines ;
- l’entrée du virus dans la cellule hôte, avec l’injection de son matériel génétique ;
- la réplication du virus dans la cellule infectée : au cours de cette étape, le virus synthétise ses protéines avec l’aide de la machinerie cellulaire de son hôte ;
- l’assemblage de ces protéines pour produire de nouveaux virions ;
- la libération des virions nouvellement formés hors de la cellule hôte, ce qui provoque l’éclatement de la cellule. Les nouvelles particules virales sont prêtes à infecter d’autres cellules pour répéter le même cycle.
Chacune de ces étapes fait intervenir des protéines spécifiques qui participent à la biologie du virus et constituent des cibles thérapeutiques potentielles. Il est donc crucial d’analyser le génome du virus afin d’identifier les gènes codant pour ces protéines.
Comparer pour prédire les gènes
L’analyse de génomes par des approches de bio-informatique est une discipline dont les principes fondateurs remontent aux années 1980-90. Il existe deux grandes approches pour identifier les gènes : l’approche ab initio et l’approche par homologie. Dans la première, on recherche systématiquement dans la séquence d’ADN des signaux universels intrinsèques qui caractérisent les portions du génome qui codent pour des protéines (voir l’article À la recherche de régions codantes). On obtient ainsi des prédictions des positions des gènes sans faire de comparaison avec d’autres génomes. L’approche par homologie, quant à elle, s’appuie sur des connaissances déjà accumulées sur des génomes apparentés. Suivant le principe de l’évolution moléculaire, un degré significatif de ressemblance entre deux séquences sert de preuve pour établir que les séquences partagent une histoire évolutive commune récente et que les éléments fonctionnels trouvés dans une séquence, comme les gènes, sont aussi présents dans l’autre séquence. En comparant les génomes avec des algorithmes d’alignement de séquences (voir l’article Comment la bio-informatique a résolu le puzzle du génome du SARS-CoV-2), on peut ainsi transférer les connaissances acquises sur des génomes bien étudiés vers un génome nouvellement séquencé.
Pour le SARS-CoV-2, la comparaison se fait avec d’autres bétacoronavirus connus. Cette recherche a révélé que le nouveau génome contient 27 protéines conservées chez les bétacoronavirus, la plupart d’entre elles étant présentes dans tous les coronavirus (voir figure 1 ci-dessous).
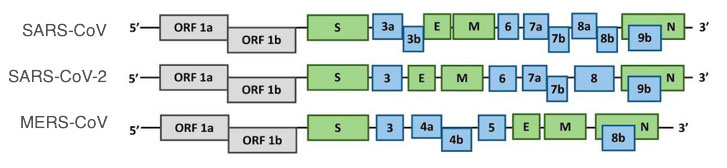
Figure 1 : Organisation du génome du SARS-CoV-2. Crédit Image : CC BY Fung et al.
L’ORF1a et l’ORF1b contiennent 16 protéines non structurelles qui sont nécessaires à la réplication et à la transcription du génome viral. Les gènes codant pour les protéines de structure spike (S), de l’enveloppe (E), de la membrane (M) et de la nucléocapside (N) sont en vert. Les gènes codant pour les protéines accessoires sont en bleu. Les génomes de deux autres bétacoronavirus humains sont également affichés pour illustrer la conservation entre des virus proches : SARS-CoV et MERS-CoV.
Plus précisément, ces protéines sont obtenues par traduction de portions du génome, les gènes correspondant à des cadres ouverts de lectures. Ce passage du génome aux protéines est explicité sur la figure 2 ci-dessous.
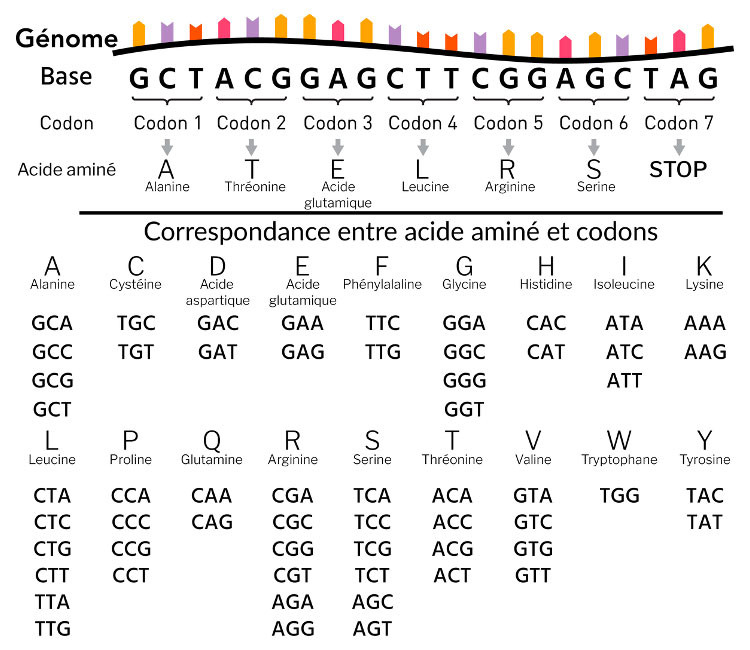
Figure 2 : la traduction et le code génétique : des gènes aux protéines.
Les séquences d’acides aminés des protéines sont dérivées des séquences génomiques par une traduction suivant le code génétique, qui associe à chaque groupe de trois nucléotides un des vingt acides aminés entrant dans la composition de la séquence protéique. Ces vingt acides aminés sont représentés par les vingt lettres A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y. Le principe de cette traduction est simple. Chaque gène démarre par un triplet de nucléotides particulier, appelé codon START. Il est ensuite composé de triplets consécutifs, correspondant chacun de manière univoque à un acide aminé, et se termine enfin par un codon STOP, le triplet TAG. Les régions ainsi délimitées par un codon START et un codon STOP dans la même phase sont appelées des cadres ouverts de lectures.
Parmi les séquences protéiques identifiées dans le génome du SARS-CoV-2, on peut trouver quatre protéines de structure qui constituent la particule virale et sont nécessaires pour que le virus infecte les cellules : la protéine spike S qui sert de médiateur à l’entrée du virus dans la cellule hôte, la petite protéine d’enveloppe E qui donne au virion sa forme définitive, la protéine de nucléocapside N qui lie l’ARN viral et interagit également avec un certain nombre de composants cellulaires, et la protéine membranaire M impliquée dans les étapes d’assemblage et de libération du virus. Bien qu’elles soient très similaires aux protéines trouvées dans d’autres bétacoronavirus, ces séquences de protéines de structure présentent plusieurs différences locales qui sont spécifiques au SARS-CoV-2 et sont probablement l’une des causes de la divergence fonctionnelle et pathogène de ce virus.
Aller plus loin avec les motifs protéiques
Pour mesurer l’impact potentiel de ces différences, on peut s’intéresser aux motifs présents dans la séquence d’acides aminés de la protéine. Des motifs sont des régions de la protéine qui sont plus spécifiquement impliquées dans la fonction et la structure de la molécule, et qui présentent un degré de conservation plus élevé entre les espèces. La figure 3 ci-dessous montre un exemple de motif, celui du domaine de liaison au récepteur (RBD) de la protéine spike S du SARS-COV-2.
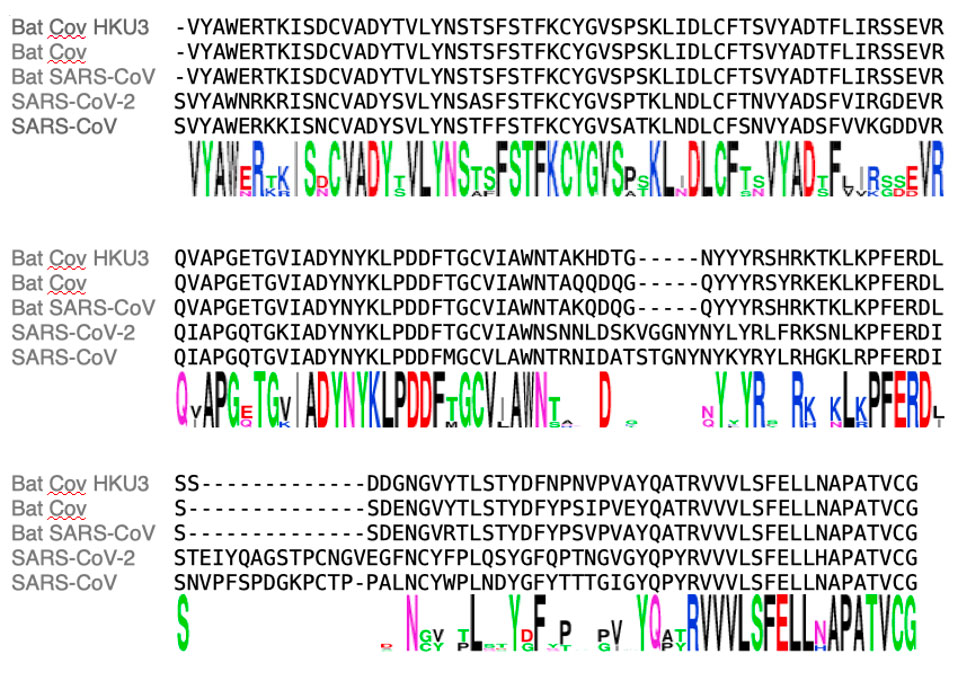
Figure 3 : Exemple de motif avec le domaine RBD de la protéine S. La séquence de la protéine S du SARS-CoV-2 fait 1310 acides aminés de long. Elle présente entre les positions 349 et 526 un motif RBD (receptor binding domain), qui se fixe au récepteur de la cellule hôte, initiant ainsi l’infection. Ce motif a été identifié par similitude avec d’autres motifs RBD dans d’autres coronavirus en utilisant un modèle de Markov à états cachés. Dans l’exemple, les trois premières séquences proviennent de bétacoronavirus de chauves-souris (Bat Cov HKU3, Bat CoV, et Bat SARS CoV). Les deux dernières séquences sont SARS-CoV-2 et SARS-CoV. La dernière ligne montre les acides aminés conservés entre les cinq séquences, à travers leur contenu en information (variant de 0 à 2 bits). Les positions les mieux conservées, retrouvées dans les cinq séquences, sont les plus hautes.
Ces motifs sont construits avec des modèles de Markov à états cachés (MMC), qui sont des modèles statistiques largement utilisés en apprentissage. Les MMC ont été introduits dans les années 1960 et s’appliquent avec succès à de nombreux domaines de l’informatique : reconnaissance des formes, traitement automatique du langage naturel, cryptanalyse, etc. Décortiquons le vocable. « Modèle de Markov » signifie que le processus à modéliser comprend plusieurs états et qu’il est sans mémoire : la valeur de chaque état à l’instant t+1 ne dépend que des valeurs à l’instant présent t. « Cachés » signifie que les états du modèle ne sont pas directement connus mais que l’on doit se fier à une série d’observations pour les deviner. Un modèle de Markov à états cachés est ainsi défini par un ensemble fini d’états dont le passage de l’un à l’autre est décrit par des probabilités, et chaque état est associé à un ensemble fini d’observations, elles aussi munies de probabilités. Dans le cas de motifs protéiques, les états sont les différentes positions du motif et les observations sont les acides aminés. L’apprentissage des états et des paramètres statistiques se fait à partir d’exemples positifs, c’est-à-dire des séquences connues pour porter la fonction concernée, et d’exemples négatifs, qui sont ici des séquences aléatoires. Un tel modèle associe à chaque position du motif les probabilités d’occurrence de chacun des différents acides aminés, d’une insertion ou d’une délétion. On peut alors calculer la probabilité qu’une portion de séquence quelconque soit générée par ce modèle et rechercher de manière automatique la présence du motif dans des séquences plus longues. La figure 4 ci-dessous détaille la construction d’un modèle de Markov à états cachés.
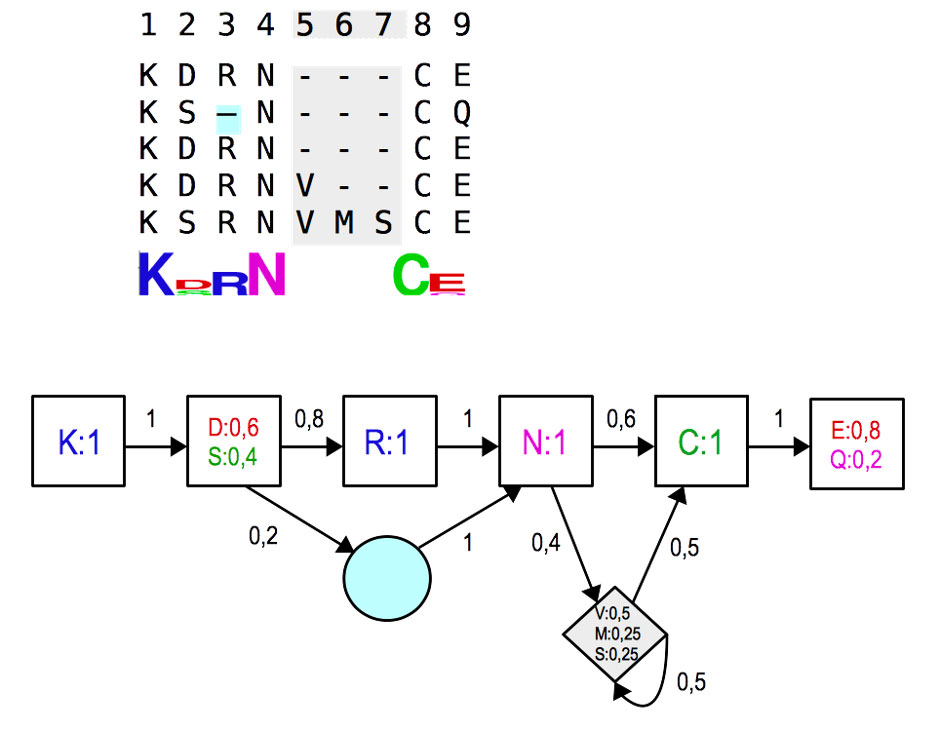
Figure 4 : Modélisation d’un motif avec un modèle de Markov à états cachés. Pour modéliser un motif, tel que le RBD, on part d’un ensemble de séquences alignées qui contiennent ce motif. À partir de l’alignement, il est possible de construire un modèle de Markov à états cachés comme suit. Les états reflètent la structure de l’alignement : les carrés sont pour les positions majoritairement conservées, les cercles pour les positions manquantes et les losanges pour les positions insérées. Sur l’exemple, il y a 6 positions conservées (colonnes 1, 2, 3, 4, 8 et 9), un caractère manquant en colonne 3 (en bleu clair sur l’alignement multiple) et une zone insérée avec les colonnes 5, 6 et 7 (en gris clair sur l’alignement multiple). Cela fait en tout un modèle à huit états. Chaque flèche entre deux états est une transition qui est associée à une probabilité. Ainsi, la probabilité de passer du premier état au deuxième est égale à 1. La probabilité de passer du deuxième état au troisième état est égale à 0,8, car 4 séquences sur 5 y vont. Enfin, chaque état est associé à des probabilités d’émission, qui correspondent à la fréquence de chacun des acides aminés. Ainsi, le premier état émet K avec une probabilité égale à 1, car toutes les séquences contiennent un K en première position. Le deuxième état émet D avec une probabilité de 0,6 (3 séquences sur 5) et S avec une probabilité de 0,4 (2 séquences sur 5), et ainsi de suite.
Pour une séquence donnée, ce modèle permet de déterminer son adéquation au motif, en calculant la probabilité de la meilleure suite d’états, acide aminé par acide aminé. La séquence KSRNCQ a ainsi une probabilité de 1 × 1 × 0,4 × 0,8 × 1 × 1 × 0,6 × 1 × 0,2 = 0,0384.
L’analyse de la séquence du génome du SARS-CoV-2 a permis d’identifier les éléments fonctionnels du virus, mettant en évidence que la structure et le contenu de ce génome n’ont rien d’exceptionnel et sont typiques d’un coronavirus. Cette étape a également permis d’identifier des portions clés du génome jouant un rôle plus important dans l’infection et la maladie. Ces portions, et en particulier les protéines associées, constituent des cibles thérapeutiques, et des séquences à surveiller plus particulièrement avec l’apparition de nouveaux variants (voir l’article « Le SARS-CoV-2 : une expérience inédite de surveillance génomique mondiale« ).
À vous de jouer !
Si le cœur vous en dit, nous vous guidons pour utiliser les outils bioinformatiques et les ressources génomiques disponibles sur le web. En quelques clics et quelques minutes, retrouvez le gène Spike S dans le génome du SARS-CoV-2 et visualisez ses similarités et différences avec ses cousins chez d’autres coronavirus.
Décodez le génome du SARS-CoV-2…
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !



Claire Lemaitre
Chargée de recherche Inria en bioinformatique au sein de l'équipe Genscale au centre Inria de Rennes.