Décoder le vivant

Deux scientifiques travaillant avec un séquenceur ADN (© Inserm/Latron P.)
Ces deux chercheuses travaillent avec un séquenceur d’ADN. Cet équipement de laboratoire est désormais incontournable pour décoder l’information génétique des organismes vivants et ainsi mieux les connaître.
Au cœur de chaque cellule existent des chromosomes qui contiennent toute l’information nécessaire à sa fabrication et à son fonctionnement : l’information génétique. Les chromosomes sont formés d’un long polymère, l’ADN (abréviation d’acide désoxyribonucléique), où les nucléotides (molécules élémentaires) se succèdent à la manière de ces colliers de bonbons dont raffolent les enfants. Une tâche essentielle des biologistes pour étudier en profondeur un organisme vivant est d’obtenir la succession de nucléotides qui sont les perles d’information de ces énormes colliers d’ADN : on appelle cette tâche « séquencer » le génome de l’organisme. On séquence en général un mélange de génomes de plusieurs individus d’une même espèce. Pour l’espèce humaine, plus de 99,5 % du génome est commune à deux individus, le reste caractérisant nos différences visibles ou la plupart du temps invisibles et se traduisant par exemple en termes de susceptibilité à des maladies.
Les techniques de séquençage de l’ADN ont bénéficié d’énormes progrès ces vingt dernières années. Obtenir la séquence de trois milliards de nucléotides du génome humain en 2003 a demandé de l’ordre d’un milliard de dollars et treize ans d’efforts, ce qui peut être obtenu aujourd’hui pour le prix et délai de livraison d’une voiture ! La disponibilité des séquences génomiques d’un nombre croissant d’organismes vivants a déjà permis de mieux connaître et comprendre la structure et l’évolution des génomes, ainsi que les mécanismes cellulaires qui exploitent l’information qu’ils portent. Au-delà, les impacts dans les domaines des biotechnologies, de l’agroalimentaire et de la santé animale et humaine, promettent d’être considérables. Disposer, par exemple, des séquences de plusieurs souches d’une même bactérie doit permettre de les caractériser en termes de pathogénicité (susceptibilité de maladie) ou d’efficacité dans des processus industriels de synthèse ou de dégradation. L’étude fine du polymorphisme humain ouvre par ailleurs la voie à une médecine personnalisée.
Un travail à la chaîne
Depuis le premier génome entièrement séquencé en 1995, celui de la bactérie Haemophilus influenzae, l’exploration des génomes se poursuit de façon systématique. Les évolutions technologiques ont permis d’accroître dans des proportions importantes et à moindre coût la quantité et la fiabilité des données recueillies. Ainsi, on disposait en mai 2009 des génomes de près de 1 000 espèces différentes auxquels il faut ajouter des génomes de communautés microbiennes entières (sources hydrothermales, appareil digestif humain…).
La séquence génomique obtenue recèle la plupart des informations déterminant le comportement d’une cellule dans un milieu donné. Une séquence est un « texte » ou plutôt une suite monotone de quatre lettres : A, C, G et T, qui distinguent les quatre types de nucléotides : adénine, cytosine, guanine et thymine. Les séquences ont des longueurs variables de quelques milliers à quelques milliards de caractères. Un biologiste possédant la séquence ADN d’un organisme vivant reste pourtant, à la simple lecture du « texte », dans l’ignorance presque complète sur sa signification. Débute alors un long travail d’interprétation, « l’annotation », consistant à repérer les morceaux de séquence correspondant à des gènes, à reconstituer les séquences de protéines synthétisées par la cellule à partir de l’information portée par ces gènes, à repérer les classes d’ARN (abréviation d’acide ribonucléique) qui régulent la machinerie génétique… Plus généralement, il s’agit d’identifier tous les objets moléculaires connus et qui sont codés par autant de morceaux de séquences dans le génome.
Ce qui restait il y a quelques années un travail de bénédictin est maintenant largement automatisé, à l’aide d’outils mis au point par les bio-informaticiens. La vérification des résultats, elle, reste manuelle : après les annotations (ou interprétations) automatiques, des erreurs demeurent, qu’il faut traquer en recoupant différentes sources d’information. Une fois validées, les annotations sont organisées au sein de bases de données et normalisées pour permettre les comparaisons entre bases différentes. Enfin, pour faciliter les recherches scientifiques, elles sont rendues accessibles au moyen de navigateurs spécialisés.
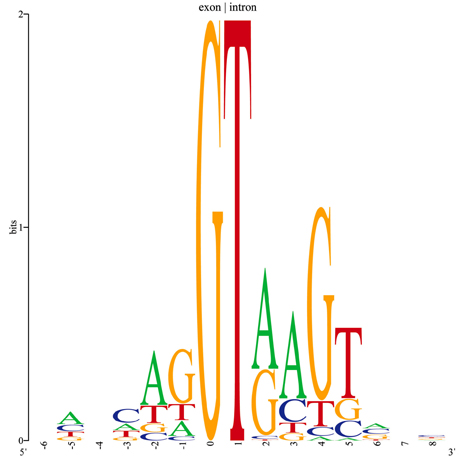
L’image décrit le motif des jonctions exon-intron dans le génome humain. Chaque gène est morcelé en une suite d’exons et d’introns alternés. Les exons sont les segments d’ADN qui codent la production d’une macromolécule active ; en revanche, les introns sont des fragments non codant. À l’aide de motifs, on peut repérer les exons et prédire le produit final d’une séquence d’ADN. Le graphique est construit à partir d’un échantillon de séquences alignées en position 0 sur un site connu de fin d’exon. À chaque position, les nucléotides apparaissent dans des proportions différentes, représentées par la taille des lettres. L’échelle verticale montre le degré de conservation d’une position. Par exemple, la position 0 est totalement conservée : toutes les séquences ont un G en 0 et la hauteur est maximale. En position 4, le G reste majoritaire mais les lettres sont plus variables, d’où une hauteur plus petite de la colonne de lettres. La hauteur se mesure en bits, unité d’information de nos ordinateurs, mais ceci est une autre histoire…
Apprendre à lire
Pour annoter, c’est-à-dire donner du sens à une séquence, le biologiste a donc besoin d’outils spécifiques. A priori, vouloir transformer une suite informe de lettres en une connaissance utile en biologie semble une entreprise désespérée. Deux faits importants vont cependant aider le bio-informaticien à extraire cette connaissance. D’une part, les séquences ne sont évidemment pas aléatoires, mais au contraire très organisées. On peut déduire de leur architecture des informations précieuses sur ce qui est codé dans la molécule d’ADN, d’ARN ou de protéine. Sur chacun des brins de la célèbre double hélice d’ADN, les nucléotides (A, C, G et T) s’organisent en mots plus ou moins longs porteurs de sens.
Par exemple, un gène de bactérie pourra se repérer comme une suite de triplets (chaque triplet détermine un acide aminé présent dans la protéine synthétisée à partir de cette région d’ADN) encadrés par des triplets spéciaux, « ATG » et « TAA », qui marquent le début et la fin des gènes, un peu à la manière des parenthèses. Le repérage n’est pas toujours facile, car un triplet qui marque le début d’une région codante peut tout aussi bien apparaître à l’intérieur d’une telle région et coder alors pour un acide aminé.
La réalité des organismes supérieurs est plus complexe et en pratique il faut retrouver des gènes, non seulement très épars dans le génome, mais aussi morcelés en une mosaïque de fragments codants et non codants qui peuvent s’assembler de différentes manières, pour produire plusieurs protéines distinctes.
D’autre part, les séquences se ressemblent d’un organisme à l’autre, et ce d’autant plus qu’ils sont proches dans l’évolution. On repère 98 % de similarité entre l’homme et le chimpanzé ! On peut donc essayer de transposer les connaissances disponibles d’une espèce à une autre, même si celles-ci sont relativement éloignées. Ainsi, travailler sur le génome de la levure de boulanger permet aussi de comprendre le génome humain. Inversement, on peut tenter de localiser un facteur de maladie en isolant la partie de génome propre à chaque individu.
Quand l’ADN se met en rang

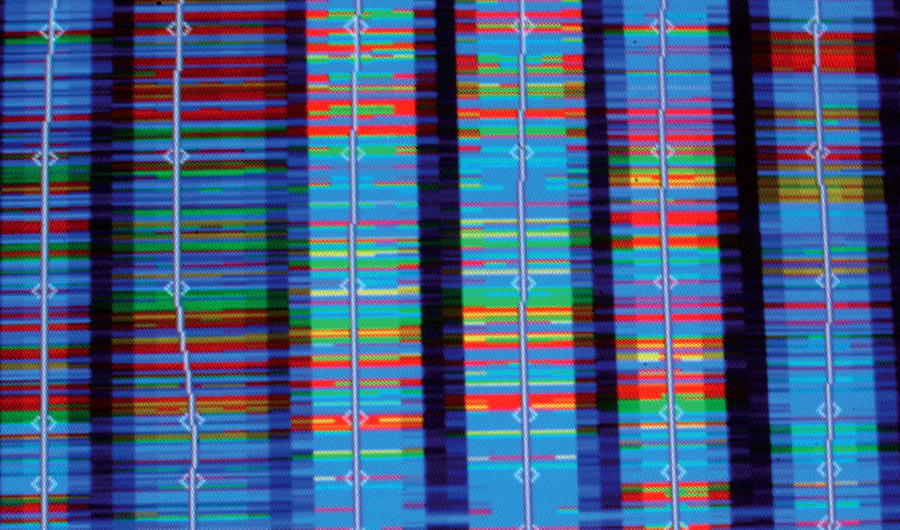
Séquençage de l’ADN (© Inserm/Depardieu M.)
Voici ce qui apparaît sur l’écran d’un séquenceur ADN : chaque colonne représente une séquence et chaque couleur une lettre de la séquence. C’est sur ces premières données informatiques brutes que le chercheur commence son décryptage.
Une série de traitements informatiques doit être appliquée aux données brutes issues d’un séquenceur pour obtenir une séquence génomique complète et de qualité. Le produit résultant est une longue chaîne de caractères (A, C, G et T), représentés ici par les différentes couleurs, qui rend compte de la succession des quatre types de nucléotides le long du brin d’ADN. Commence alors un délicat travail d’annotation de ce texte. Il s’agit d’identifier et de caractériser des régions d’intérêt, en premier lieu les gènes qui contiennent l’information nécessaire à la synthèse d’une ou plusieurs protéines. Fort heureusement, la chaîne de caractères est un objet bien connu des informaticiens pour lequel ils disposent d’un très riche arsenal d’algorithmes d’analyse, à partir desquels les bio-informaticiens ont développé des méthodes d’annotation pertinentes et efficaces. La comparaison avec des séquences déjà annotées et disponibles dans des bases de données constitue par ailleurs une source d’informations très utiles.
D’un point de vue informatique, retrouver des séquences qui se ressemblent implique des comparaisons de séquences et repose principalement sur ce qu’on appelle un « alignement ». Un alignement de séquences suppose que celles-ci sont des variations d’une même séquence d’origine (qu’avait par exemple un ancêtre commun) qui a été transformée. On peut imaginer ici chaque séquence comme une suite de perles colorées reliées par un fil élastique : un alignement consiste à tirer plus ou moins sur chaque élastique, de sorte que toutes les séquences soient de même longueur et qu’à chaque position, les perles soient de même couleur ou d’une couleur la plus similaire possible. Les molécules associées partagent des propriétés physicochimiques qui peuvent servir de base à cette comparaison. Chaque correspondance et chaque extension de fil nécessaire reçoit un score traduisant la similarité observée. La difficulté est de trouver la meilleure somme de scores possible par rapport à tous les alignements imaginables.
On dispose d’une méthode pour résoudre ce problème d’optimisation, la « programmation dynamique ». Celle-ci consiste à remplir une matrice de scores partiels dont le nombre de lignes et de colonnes est égal à la longueur des deux séquences à comparer. Mais une telle taille devient vite ingérable et il faut se contenter de méthodes moins précises mais plus rapides. Le logiciel le plus utilisé en bioinformatique, Blast, recherche au préalable des « graines », des mots qu’on peut retrouver à l’identique dans toutes les séquences à comparer. En effet, deux séquences proches ont forcément en commun des mots répétés.
Prenons une séquence constituée de 15 « A » successifs ayant subi 2 mutations au hasard : dans tous les cas, le mot « AAAAA » sera présent et on pourra trouver au moins 3 exemplaires distincts du mot « AAA ». Mieux, on peut même affirmer que la sous-séquence « AA ??A ??AA », où le « ? » indique un caractère quelconque, sera conservée. On se sert de ces mots facilement repérés et stockables dans un « dictionnaire » comme points d’ancrage pour des recherches plus complètes.
Répétition générale
Cette idée de répétition correspond aussi à une réalité biologique : les génomes contiennent un nombre élevé de répétitions de types très variés. Celles-ci jouent un rôle majeur dans la structure, la fonction et l’évolution des génomes. Leur étude repose presque toujours sur la construction d’un dictionnaire de tous les mots présents dans la séquence étudiée. En pratique, les mots d’un génome entier peuvent ainsi tenir dans la mémoire d’un ordinateur. On peut ensuite modéliser la nature des répétitions.

Maïs indien (© Fentress S.)
Dans tous les génomes, on observe des répétitions consécutives appelées « répétitions en tandem », dont on analyse la variation. Par exemple, on trouve sur notre chromosome X une suite de répétitions du triplet « CAG » de 9 à 32 fois dans le gène du récepteur aux androgènes (fixateurs d’hormones mâles). Lorsque le nombre de « CAG » dépasse 36, cela peut déclencher une maladie neurologique, la maladie de Kennedy. D’autres répétitions, les « transposons », sont capables de se déplacer et de se multiplier dans les génomes (elles forment 45 % du génome humain). En effet, le génome est une entité dynamique dont certains fragments vont s’autoexciser à certaines positions pour aller s’insérer ailleurs. Ces éléments mobiles expliquent la coloration surprenante de certaines plantes (comme le maïs présenté ci-contre).
Deux types de techniques permettent d’analyser les répétitions : soit des techniques statistiques, où on compte le nombre de fois où apparaissent certains mots pour détecter des portions anormales ou exceptionnelles du génome, soit des techniques combinatoires où on dispose d’un ensemble de contraintes à respecter pour lesquelles on énumère l’ensemble des solutions possibles.
Le génome, un tissu à motifs
Au-delà des répétitions, il existe de multiples motifs dans les séquences, structures plus ou moins complexes qui vont délimiter les éléments des gènes, engendrer des interactions entre macromolécules ou encore contraindre la localisation ou la conformation des protéines dans la cellule. L’annotation se basera sur ces motifs pour donner du sens aux séquences.
Il existe une vaste panoplie de programmes pour la découverte de motifs à partir d’ensembles de séquences qui autorisent des études plus systématiques et plus précises. Des banques de données rassemblent les motifs trouvés afin qu’ils puissent être réutilisés dans l’annotation. Deux types d’approches sont disponibles : probabilistes ou combinatoires. Les réseaux de Markov vont ainsi cribler la suite des lettres d’une séquence à travers une série de tamis où la probabilité de passage dépend de la lettre considérée. Ces probabilités sont ajustées à partir de séquences de référence déjà annotées. L’autre approche tire ses racines de la théorie des langages formels, socle mathématique des langages de programmation. Ainsi, un grand nombre de motifs comme celui des toxines se rattachent à ce que l’on appelle les « expressions régulières », pour lesquelles on dispose de programmes de recherche efficaces largement utilisés en informatique (notamment pour le traitement de textes).
La plupart des toxines de serpents, protéines responsables de la toxicité de leur venin, sont repérables dans leur séquence par un motif similaire.
Ce motif s’écrit : GC-x(1,3)-CP-x(8,10)-CC-x(2)-[PDEN], les lettres G, C, P, D, E, et N étant les initiales des acides aminés qui se succèdent. Cela signifie qu’il faut chercher une séquence avec le mot « GC » suivi de 1 à 3 lettres quelconques, du mot « CP » suivi, 8 à 10 lettres plus loin, du mot « CC », de 2 lettres et enfin d’une lettre au choix parmi P, D, E ou N. Les « C » codent pour des cystéines qui vont établir des liaisons chimiques entre elles et être responsables du blocage des commandes musculaires. De tels motifs peuvent être construits plus ou moins automatiquement, à partir des alignements d’un ensemble de séquences de référence d’une même famille.
Dans le domaine de l’analyse des séquences, les défis restent nombreux. Une prochaine étape de la conquête des génomes se pose d’ores et déjà de manière aiguë : c’est par milliers qu’il faudra désormais être capable de comparer les génomes entre eux, afin d’élucider les variations génétiques et de les mettre en correspondance avec les variations visibles au sein d’une population.
Pour en savoir plus, un site web : La foire aux questions du Génoscope.
Cet article est paru dans la revue DocSciences n°8 Le numérique et les sciences du vivant, éditée par le CRDP de l’Académie de Versailles en partenariat avec l’Inria.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

Jacques Nicolas