
Évolution et phylogénie
Si la vie n’existait pas il y a quatre ou cinq milliards d’années, aujourd’hui elle est presque partout présente, des sources chaudes à plus de 100 °C jusqu’aux glaces éternelles (enfin presque éternelles !) et sous des formes qui vont de la cellule isolée d’une bactérie jusqu’au baobab ou à la baleine. L’information génétique est au cœur du processus évolutif. Une révolution technologique, commencée au début des années 1970, fournit des données de plus en plus riches sur sa structure, son rôle et son évolution. La gestion et l’exploration de ces données ont joué un rôle moteur dans le développement de la bio-informatique, devenue un partenaire indispensable de la biologie. Aujourd’hui, un des enjeux majeurs est de comprendre comment les organismes vivants ont évolué depuis leur ancêtre commun Luca (acronyme de last universal common ancestor).
Il existe entre cinq et cinquante millions d’espèces différentes ! Cette incroyable diversité résulte de l’évolution du vivant pourtant issu d’un ancêtre commun. On retrouve son empreinte dans le code génétique qui est quasi universel.
Le vivant se caractérise par une extraordinaire diversité, comme le montre cette collection de papillons conservée au Muséum national d’histoire naturelle. Le nombre exact d’espèces différentes est loin d’être connu ; sans compter les bactéries, les estimations varient de cinq à cinquante millions ! Seules moins de deux millions sont effectivement recensées et deux tiers sont des insectes. Cette variété résulte d’une histoire évolutive qui s’étale sur plus de quatre milliards d’années. Simultanément, l’existence d’un ancêtre commun unique se traduit par une unité du vivant, dont une première manifestation est la quasi-universalité du code génétique. De même, l’observation de ressemblances entre espèces traduit, phénomènes de convergence mis à part, l’existence d’une ascendance commune plus ou moins récente. Cette filiation des espèces est à la base de la classification du vivant. Au sein même d’une espèce, les individus présentent une diversité qui résulte des mutations et pour les organismes sexués du brassage de l’information lors de la formation des gamètes.
Les bases de la génétique
L’information génétique utilise quatre motifs chimiques, appelés « bases » et dénotés par les lettres A, C, G, T. Par exemple, chez l’homme, ce sont plus de trois milliards de bases qui s’organisent en quarante-six chromosomes et constituent ainsi le génome. Les bases successives sont liées chimiquement pour constituer un polymère, le brin d’ADN. De plus, il existe des affinités particulières entre des bases : A s’apparie avec T et C avec G. Cette complémentarité permet aux chromosomes d’adopter une structure particulière dans laquelle deux brins sont appariés pour constituer une double hélice. Les deux brins complémentaires contiennent la même information. Cette redondance est centrale dans les processus de réplication de l’information et de contrôle de sa validité : chaque cellule d’un organisme contient un exemplaire du génome. Quand une cellule se divise, le génome doit être répliqué : les deux brins se séparent, de nouvelles bases viennent s’apparier à ces deux brins libres pour former deux nouveaux brins, complémentaires des brins initiaux. Ceci permet la constitution de deux nouvelles doubles hélices identiques à la première.
Un mammifère est constitué de plusieurs dizaines de billions (1 billion = 1012) de cellules (de l’ordre de 1014 chez l’homme). Toutes ces cellules proviennent par division cellulaire d’une seule cellule, l’oeuf (ou zygote), et chacune d’entre elles a nécessité une réplication du génome. L’origine de l’information génétique portée par le zygote est plus complexe, sa provenance est en effet double puisqu’elle implique les deux parents (pour simplifier, nous ne parlerons pas ici des étranges sexualités des microbes). Chaque individu porte deux copies de l’information génétique, l’une venant de sa mère et l’autre de son père. Pour participer à la génération suivante, il fabrique des gamètes qui sont des cellules très particulières, les seules qui ne contiennent qu’une seule copie de l’information génétique. Cette copie unique est constituée d’un mélange, au hasard, de fragments provenant des deux copies de l’individu. La fusion d’un gamète mâle et d’un gamète femelle reconstitue une cellule avec deux copies. Le décor est maintenant planté, l’information génétique est décrite, de même que sa propagation à l’ensemble des cellules d’un individu d’une part et aux descendants de cet individu d’autre part. La question majeure peut à présent être posée : comment cette information a-t-elle évolué et comment évolue-t-elle encore actuellement ?
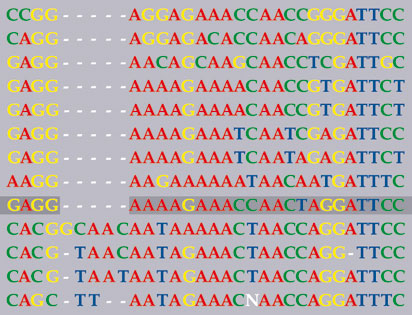
Alignement de séquences ADN (© DocSciences/Métayer A.)
En alignant, de manière à trouver le plus de similitudes entre les lettres de séquences ADN d’espèces proches, on repère des changements : apparition ou disparition d’une lettre, substitution d’une lettre par une autre… On y « lit » l’évolution.
Les génomes des différentes espèces se ressemblent, mais ne sont pas identiques. Depuis leur divergence de leur ancêtre commun, les génomes accumulent des différences qui peuvent être, soit des substitutions d’une base par une autre, soit des insertions de bases, soit des suppressions. Ainsi, si on écrit les génomes les uns au-dessous des autres, les lettres d’une même colonne ne se correspondent pas. Des algorithmes ont été développés pour mettre des insertions ou des délétions aux « meilleurs » endroits de façon à obtenir un alignement optimal, dont le coût global, somme des coûts unitaires des insertions/ délétions et des substitutions, est le plus faible possible. Le problème biologique est de fixer les coûts unitaires ; le problème informatique est de calculer des solutions optimales en un temps raisonnable. Pour des coûts de forme simple et pour deux séquences, des méthodes très efficaces existent ; pour des coûts plus complexes et des séquences en grand nombre, il n’existe pas de méthodes exactes et efficaces.
L’erreur : un des moteurs de l’évolution
Si la réplication de l’information pendant la production des gamètes était parfaite, cette information ne se modifierait pas et donc n’évoluerait pas. Si au contraire la réplication comportait de nombreuses erreurs, le texte perdrait son sens et l’organisme n’y survivrait pas. Entre ces deux extrêmes s’est construit, au cours des milliards d’années de l’évolution biologique, une situation intermédiaire dans laquelle l’existence d’erreurs, les mutations, permet une diversité autorisant l’apparition d’informations favorables, le taux d’erreurs restant suffisamment bas pour que l’existence d’informations inadaptées ne constitue pas un fardeau trop lourd pour les populations.

Charles Darwin (1809-1882) en 1881 (© Heritage Images/Leemage)
Ce qui apparaît extraordinaire est que ce modèle correspond à celui proposé il y a cent cinquante ans par Charles Darwin, alors qu’il ne connaissait rien de la notion d’information génétique. Malgré les progrès considérables des connaissances en biologie depuis la parution de son livre L’Origine des espèces, tous les faits, observations, résultats expérimentaux récents s’intègrent parfaitement à la vision évolutive de Darwin.
Ce modèle définit deux étapes : la production des mutations qui conduit à une diversité génétique, puis l’évolution de la fréquence de ces mutations au cours des générations successives. La fréquence d’une mutation augmente si elle est transmise à de nombreux individus de la génération suivante. Ainsi, si une mutation augmente la probabilité de son porteur d’avoir de nombreux descendants, sa fréquence va croître. On dira que cette mutation est « favorable » et qu’elle est « sélectionnée positivement ». Elle pourra même se « fixer » dans la population en devenant présente dans tous les génomes de cette population. Si au contraire une mutation conduit à diminuer le nombre de descendants de ses porteurs, elle sera « défavorable », sa fréquence décroîtra jusqu’à son élimination. Il est aussi possible que la mutation n’ait aucun effet sur les capacités reproductives des individus qui la portent, on parle alors de « mutations neutres ».
Reconstruire l’histoire des espèces
Un apport important des mathématiques a été de montrer que les mutations neutres se fixaient dans les espèces à un rythme régulier et contribuaient ainsi à créer un pourcentage de différences entre les génomes des espèces proportionnel au temps écoulé depuis leur divergence de leur plus proche ancêtre commun. Il devenait ainsi possible de reconstruire l’histoire des espèces en déduisant l’arbre phylogénétique des distances entre séquences génomiques. Mathématiques et informatique ont collaboré pour concevoir les modèles et les algorithmes permettant cette reconstruction.
Un arbre phylogénétique représente les relations de parenté qu’un ensemble d’espèces entretiennent de par leur histoire évolutive commune. Les feuilles de l’arbre, ou nœuds terminaux, sont associées aux espèces actuelles ; le plus récent ancêtre commun à deux espèces est associé au nœud qui connecte ces dernières ; et ainsi de suite, jusqu’à la racine de l’arbre phylogénétique. Chaque gène contribue ainsi à la compréhension de l’histoire de la vie, cependant les histoires racontées par chacun d’eux sont parfois différentes et c’est souvent de ces différences que les résultats scientifiques les plus intéressants ont émergé. Il a ainsi été mis en évidence que les gènes peuvent voyager entre espèces différentes. Ces « transferts horizontaux » jouent un rôle évolutif important, les capacités de certaines espèces pouvant alors être acquises par d’autres. Ainsi s’explique par exemple la résistance aux antibiotiques chez certaines bactéries.
Un arbre phylogénétique est une reconstruction de l’histoire des espèces. Cependant, cette structure ne rend pas compte des transferts horizontaux : un fragment génomique d’une espèce peut être directement intégré au génome d’une autre espèce. De multiples mécanismes biologiques peuvent réaliser de tels transferts. Ainsi les bactéries disposent d’une panoplie complexe de vecteurs capables d’assurer la dissémination, entre espèces, de gènes de résistance aux antibiotiques. Les virus peuvent à la fois incorporer dans leur génome des gènes de l’hôte et intégrer tout ou partie de leur génome dans celui de la cellule qu’ils infestent et ainsi occasionner parfois de sévères dysfonctionnements. La prise en compte, difficile, de ces transferts horizontaux a tendance à transformer les arbres en graphes, autre structure que les informaticiens affectionnent…
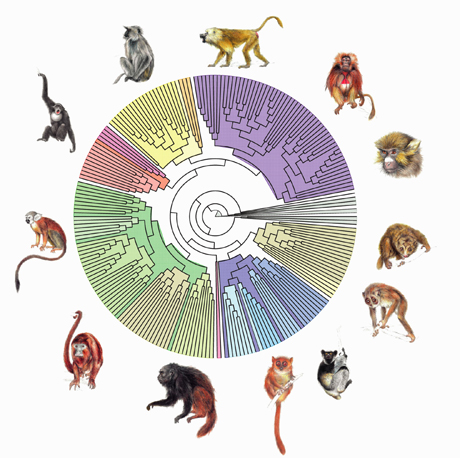
Arbre phylogénétique des primates (© LIRMM/CNRS/Gascuel O., Guindon S. – ISEM/CNRS/Douzery E., Fabre P.-H. – IRD/Chevenet F.)
Un arbre phylogénétique reconstruit l’histoire des espèces. Par exemple, ici, en partant des feuilles, représentées par les singes, on peut retracer, grâce à un logiciel comparant les séquences ADN, la filiation jusqu’à l’ancêtre commun à tous les primates.
Cet arbre phylogénétique incorpore 217 des 233 espèces actuelles de primates. Il a été reconstruit grâce au logiciel PhyML, utilisant un puissant algorithme, mis au point en 2003, qui permet d’estimer les relations évolutives liant un ensemble d’organismes à partir de la comparaison de séquences d’ADN représentant plus de 900 000 lettres. Les feuilles de l’arbre, ici figurées par les dessins de primates à la périphérie, représentent les espèces actuelles, les nœuds correspondent aux espèces ancestrales et les branches retracent les temps évolutifs séparant les espèces. On retrouve les Lorisiformes en brun, les Lémuriens en bleu, les Tarsiers en rose, les Platyrrhiniens en vert, les Hominoïdes en rouge, et les Cercopithécoïdes en jaune et violet. Aujourd’hui, grâce à de tels algorithmes, sans cesse en cours d’amélioration, les temps de calcul sont de plus en plus réduits, et plus rien n’empêche d’aborder des recherches mettant en jeu 500 espèces et des séquences de plusieurs dizaines de milliers de lettres !
Une caractéristique majeure du modèle est l’indépendance entre la production de mutations et la sélection de celles-ci : la probabilité d’apparition d’une mutation n’a pas de relation avec son statut favorable, défavorable ou neutre. On énonce souvent cela de manière assez malheureuse en disant que les mutations se produisent « au hasard ». En fait, une mutation résulte à la fois d’une erreur et de l’effet de multiples processus de réparation des erreurs, c’est donc un phénomène complexe ayant une forte composante biologique. Son impact évolutif est important, car c’est lui qui définit le mode d’exploration de l’ensemble, quasiment infini, des informations théoriquement possibles.
Le processus mutationnel : une histoire sans fin
Ainsi, certaines caractéristiques des génomes sont dues seulement à des « biais » dans les mutations. Un des plus célèbres biais mutationnels est la très faible fréquence de C suivi d’un G dans les génomes, par exemple, de mammifères. Ceci résulte d’une transformation chimique fréquente des C en T. Par contre, la suite TG est anormalement fréquente. D’autres caractéristiques des processus mutationnels, bien qu’indépendants de la sélection, favorisent les processus d’adaptation. En voici deux exemples de nature différente.
Le premier a été mis en évidence chez les bactéries. Lorsqu’une population bactérienne est soumise à des changements très fréquents d’environnement, des formes défectueuses de gènes nécessaires à la réparation des erreurs sont sélectionnés, entraînant une augmentation du taux de mutation. Cela conduit à une grande diversité génétique dans cette population et augmente ainsi la probabilité d’une configuration génétique favorable. Bien entendu, il existe un coût et cette diversité n’est maintenue par la sélection que si le changement rapide du milieu impose une constante adaptation de l’information génétique.
Le deuxième exemple provient de deux observations. D’une part, un type de mutation fréquente conduit à la duplication ou à la suppression de fragments génomiques de grande taille, et d’autre part, l’information génomique est modulaire, et ceci à plusieurs échelles. Par exemple, les informations relatives à la structure d’une protéine et celles concernant les conditions requises pour qu’elle soit fabriquée sont écrites séparément, il est donc « facile » de modifier la production d’une protéine sans en changer la structure. Par ailleurs, la structure même d’une protéine est très souvent organisée en régions ayant des rôles fonctionnels identifiés, et on remarque que ces régions sont souvent partagées entre des protéines différentes. Ainsi les mutations capables de « brasser » des fragments du génome, associées à la modularité des protéines, apparaissent comme un puissant moteur de création de protéines ayant des activités nouvelles. Il reste à démontrer que ces mécanismes mutationnels ont été sélectionnés pour permettre une exploration efficace des possibles, en particulier en facilitant la réutilisation de fragments d’information déjà « optimisés ».
Un des enjeux majeurs de la biologie est de construire un lien cohérent de l’échelle de la molécule jusqu’à celle des populations et des communautés d’espèces différentes. L’analyse au niveau moléculaire des mécanismes d’expression de l’information génétique et des mécanismes de génération de la diversité biologique doit être associée à celle des mécanismes d’adaptation des organismes à leur environnement et aux changements de cet environnement. Les mathématiques et l’informatique joueront un rôle de plus en plus important dans cet effort d’intégration de la biologie moléculaire à l’écologie. La gestion des connaissances doit permettre aux biologistes d’intégrer les concepts des autres champs de la biologie. Les modèles sont l’outil indispensable pour mettre en évidence les interactions possibles entre diverses composantes de systèmes biologiques complexes et multi-échelles.
- Darlu P., Tassy P., La Reconstruction phylogénétique. Concepts et Méthodes, Masson, 1993, en téléchargement sur la page Web : Publications de la Société Française de Systématique.
- Lecointre G., Le Guyader H., Classification phylogénétique du vivant, Belin, 2006.
- Lecointre G. (ss dir.), Comprendre et enseigner la classification du vivant, coll. « Guide Belin de l’enseignement », Belin, 2008.
- Le Guyader H., Classification et évolution, coll. « Le collège de la cité », Le Pommier, 2003.
- Mayr E., Après Darwin. La biologie, une science pas comme les autres, coll. « Quai des sciences », Dunod, 2006.
- Perrière G., Brochier C., Concepts et méthodes en phylogénie moléculaire, coll. « Iris », Springer, 2009.
Cet article est paru dans la revue DocSciences n°8 Le numérique et les sciences du vivant, éditée par le CRDP de l’Académie de Versailles en partenariat avec l’Inria.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

Christian Gautier
Professeur à l’université Lyon 1, laboratoire de Biométrie - Biologie évolutive (UMR CNRS 5558).