La molécule sur écran
La fonction d’une protéine est en grande partie liée à la structure que sa chaîne polypeptidique (plusieurs acides aminés) acquiert lors de sa maturation. C’est cette conformation spatiale (manière dont la chaîne se replie dans l’espace) qui va, par exemple, dicter la spécificité d’une éventuelle activité catalytique. Or, la détermination expérimentale de la structure d’une protéine est un processus encore long et délicat. Aussi, dès qu’ils ont pu disposer des premières séquences polypeptidiques, les bio-informaticiens ont cherché à prédire la structure d’une protéine à partir de la seule donnée de sa séquence. Simultanément, ils ont développé des algorithmes pour la comparaison, la visualisation et la manipulation directe de ces structures moléculaires.

Maquette de la molécule d’ADN (© Inserm/Depardieu M.)
Cette maquette réelle représente une molécule en trois dimensions. Aujourd’hui, grâce à l’informatique, on reproduit non seulement des molécules virtuelles, mais on peut également les manipuler, les animer, simuler leur évolution…
Cet assemblage de boules de plastique reliées par des tiges de fer est l’une des premières représentations moléculaires en trois dimensions. Ici, c’est une représentation de la structure de l’ADN en double hélice ? identifiée par Watson et Crick en 1953. La tige centrale (support) correspond à l’axe de l’hélice. Les boules correspondent aux atomes. Leurs couleurs indiquent la nature des atomes (oxygène en rouge, carbone en noir, azote en bleu, phosphore en mauve et hydrogène en blanc). Les tiges de fer reliant les atomes représentent les liaisons covalentes. Les bases (au centre) des deux brins antiparallèles (extérieur) sont appariées lors de la formation de la double hélice par des liaisons hydrogène. On distingue une asymétrie dans l’hélice avec un grand sillon visible sur la gauche. De nombreux composés antitumoraux interagissent avec l’ADN dans ce sillon. La structure de l’ADN n’est pas figée comme suggéré par ce modèle. La structure et la courbure de l’hélice sont affectées, notamment lors d’interactions avec des protéines dans le cadre de la régulation de l’expression génétique.
Des séquences, des structures, des fonctions
Une macromolécule biologique, qu’il s’agisse d’un acide nucléique (ADN, ARN) ou d’une protéine, est le plus souvent représentée par sa séquence, une représentation linéaire, à une seule dimension. L’information génétique est ainsi codée dans les séquences d’ADN, qui sont transcrites en séquences d’ARN, elles-mêmes traduites en séquences d’acides aminés pour synthétiser les protéines.
Les protéines synthétisées adoptent ensuite une conformation tridimensionnelle spécifique, qui implique des proximités spatiales entre groupements chimiques distants dans la séquence. Cette conformation a des propriétés physiques et chimiques particulières qui sont le support de l’activité biologique de chaque protéine. Il s’établit donc une relation entre la séquence d’une protéine, sa structure et sa fonction.
La taille des protéines est de l’ordre de quelques dizaines à plusieurs centaines d’angströms (un dix-milliardième de mètre, 1 Å = 10 – 10 mètre). Cette taille est hors du champ d’investigation des techniques optiques. Déterminer la structure spatiale d’objets aussi petits se fait donc par des techniques expérimentales couplées à des méthodes de calcul informatisées.
Plusieurs méthodes expérimentales sont actuellement disponibles. La radiocristallographie aux rayons X bombarde un cristal de protéine par un faisceau de rayons X. C’est l’analyse des images de diffraction obtenues qui permet de calculer les positions des atomes dans le cristal. La résonance magnétique nucléaire (RMN) mesure les proximités entre certains atomes. On obtient ainsi des distances entre paires d’atomes. À partir de ce jeu de données, il est possible de reconstruire la structure d’une protéine. Enfin, la microscopie électronique offre une précision moindre. Elle permet dans certains cas de visualiser une enveloppe. On essaie alors de calculer une structure capable d’entrer dans cette enveloppe.
La première structure tridimensionnelle d’une protéine a été obtenue à partir de données de radiocristallographie électronique. Les calculs nécessaires à l’interprétation des données expérimentales ont demandé plusieurs années d’efforts à John Kendrew et ses collaborateurs pour proposer, en 1958, un modèle structural de la myoglobine (protéine présente chez les vertébrés, proche de l’hémoglobine). Le développement de programmes informatiques complexes a depuis lors permis d’automatiser le traitement des données expérimentales et de réduire ainsi le temps de traitement à quelques heures.
Un œil sur la structure des protéines
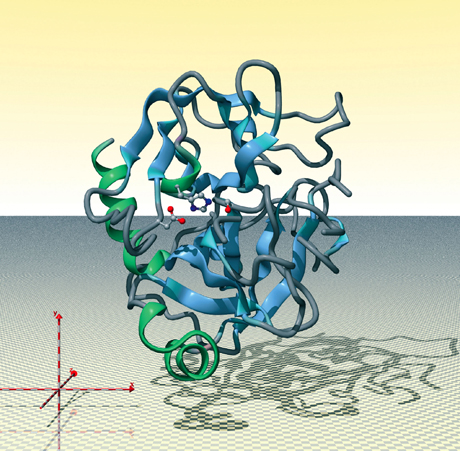
Chymotrypsine, image générée par le Protein Movie Generator (© DR)
Voir une protéine en trois dimensions permet d’explorer sa fonction. En effet, les chercheurs ont mis en évidence une relation entre la structure d’une protéine, la manière dont sont organisés ses composants chimiques et son rôle dans l’organisme.
Dans la structure d’une protéine, la proximité spatiale d’acides aminés distants dans la séquence confère des propriétés fonctionnelles spécifiques. La chymotrypsine est une enzyme digestive fabriquée par le pancréas. Elle participe à la dégradation des protéines absorbées. Cette représentation de la protéine montre le squelette (trame de la polymérisation des acides aminés) sous une forme de ruban avec, en son centre, les trois acides aminés, de gauche à droite : aspartate, histidine, serine. Ils sont impliqués dans l’activité catalytique de l’enzyme et sont voisins dans la structure, mais répartis dans la séquence sur plus de 130 acides aminés. Les couleurs utilisées pour les atomes sont le rouge pour l’oxygène, le bleu pour l’azote, le gris pour le carbone. De nombreuses enzymes ont une telle triade impliquée dans leur activité catalytique. La variation du type et de la position dans l’espace des acides aminés confère à chaque type d’enzyme une activité particulière.
La visualisation de la structure des protéines est d’une grande aide pour comprendre les mécanismes fonctionnels dans lesquels elles sont impliquées. En effet, voir les structures, c’est accéder à la compréhension des proximités spatiales. Et la proximité spatiale d’acides aminés distants dans la séquence confère à la protéine des propriétés fonctionnelles spécifiques. Dans une enzyme comme la chymotrypsine, ce sont trois acides aminés distants de plus de 100 positions dans la séquence, mais qui sont proches dans l’espace, qui sont responsables de l’activité catalytique.
L’exploration visuelle des structures protéiques n’est pas simple. Tout d’abord, le nombre d’atomes peut atteindre plusieurs milliers, voire plusieurs dizaines de milliers. Comment aller à l’essentiel ? Quelle représentation choisir pour visualiser l’information pertinente contenue dans les structures ? C’est l’objet de l’infographie moléculaire.
Modèle obtenu par superposition à l’aide d’un système de visualisation (© Inserm/Roques B.)
Les premiers modèles structuraux ont été construits comme des maquettes matérielles, à l’aide de tiges de fer reliant les positions des atomes liés. Évidemment, c’était long, fastidieux et fragile. C’est au milieu des années 1960 que remonte la première visualisation d’une molécule sur un écran. La visualisation informatique, ou graphisme moléculaire, s’est ensuite rapidement imposée. Elle a posé deux grands défis : dessiner sur l’écran et interagir avec celui-ci. Les premiers écrans graphiques, dits calligraphiques, étaient proches d’un oscilloscope. Ils permettaient de tracer des segments de droites entre deux points quelconques de l’écran. La représentation des molécules consistait alors en un ensemble de segments de droites représentant les liaisons covalentes entre atomes.
Les écrans graphiques à balayage de trame – similaires aux écrans actuels – ne datent que des années 1980. Leur arrivée a permis un grand pas qualitatif dans le réalisme de la visualisation : dessiner des surfaces, gérer la transparence d’objets, modéliser leur éclairage, les ombres… Un plus grand nombre de modes de représentations sont disponibles : surfaces, représentations simplifiées à base de splines (fonctions mathématiques utilisées pour créer des contours complexes)… Il devient surtout possible de combiner différents types de représentations, par exemple en schématisant la trame du repliement, tout en affichant explicitement les atomes impliqués dans des acides aminés particuliers.
Mettre les molécules en mouvement
Comment manipuler un objet sur un écran pour l’examiner de plusieurs côtés ? Faire tourner les molécules sur un écran a demandé le développement d’un formalisme mathématique utilisant des coordonnées homogènes. Ce formalisme, qui date des années 1970, permet la combinaison des translations et des rotations par des opérations matricielles. Ce type d’opérations est actuellement pris en charge en grande partie par les cartes graphiques, ce qui améliore l’interactivité. Désormais, l’infographie moléculaire permet de générer des animations illustrant les aspects dynamiques des structures ou l’interaction entre molécules.
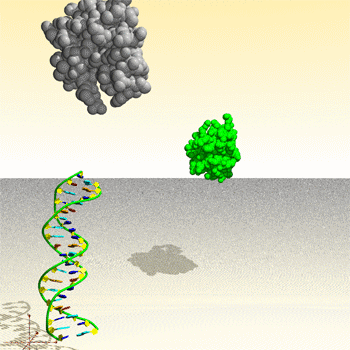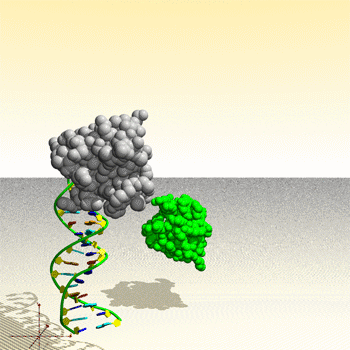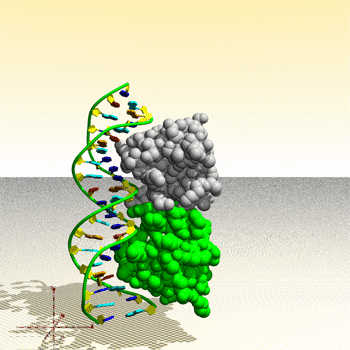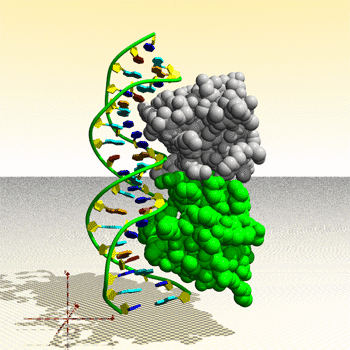
Assemblage de complexe ADN – protéine : image générée par le Protein Movie Generator.
Rendre possible la comparaison croisée à grande échelle des séquences et des structures, c’est sans doute l’apport le plus important de l’outil informatique à la compréhension de la structuration en trois dimensions des protéines. L’infographie moléculaire a vite montré que certaines protéines ont des structures voisines. Comment mesurer la ressemblance entre deux structures ? Tout comme la recherche de similitudes entre les séquences se fait en termes d’alignement, c’est-à-dire de la mise en concordance des acides aminés sur la base de leurs similitudes physico-chimiques, on peut aussi superposer des structures et déduire un alignement structural sur la base des appariements 3D. Un résultat fondamental a été de constater que des protéines de séquences voisines se replient généralement d’une façon similaire : une séquence proche donne une structure tridimensionnelle proche, mais la réciproque n’est pas vraie. Si l’on connaît la structure d’une protéine suffisamment proche, homologue, de celle que l’on souhaite étudier, on peut calculer, modéliser, sa structure.
Cette constatation a donc permis de s’affranchir des données expérimentales pour modéliser à grande échelle la structure des protéines. En conséquence, un effort particulier a été fait pour déterminer par des techniques expérimentales la structure de protéines de séquences non homologues à celles de structure connue. Les données s’accumulant, un autre résultat a été de constater que le nombre de façons dont les protéines se replient semble limité. À beaucoup de séquences différentes correspondent peu de familles structurales.
La connaissance des structures n’est pas tout. Les molécules ne sont pas rigides. Les atomes qui les constituent interagissent et bougent. Les structures subissent des déformations à différents niveaux. De plus, les molécules ne sont pas isolées, mais interagissent avec un solvant et d’autres macromolécules ou constituants cellulaires. Ces fluctuations conformationnelles des molécules sont explorées par la simulation moléculaire, basée sur l’intégration numérique des équations de Newton. Là encore, cette problématique n’est pas nouvelle. Les premiers essais de simulation moléculaire sur ordinateur datent de la fin des années 1950. Mais la simulation est très coûteuse en temps de calcul. Pour qu’elle ait un sens physique, il faut calculer les déplacements de chaque atome chaque femtoseconde (1 Fs = 10 – 15 s) ! Ceci justifie l’emploi de puissants calculateurs parallèles. Actuellement, des fluctuations conformationnelles peuvent être simulées sur une période maximale de l’ordre de la microseconde (1 µs = 10 – 6 s). Cette échelle de temps permet d’observer certaines transitions importantes pour la fonction des protéines, leurs interactions. Mais c’est encore trop peu pour simuler le repliement des protéines qui dure plus d’une milliseconde. La simulation moléculaire a donc besoin d’ordinateurs plus performants.
Une application : la recherche de médicaments
La conception d’un médicament est un processus long (environ quatorze ans) et coûteux (800 millions d’euros). Là encore, l’informatique apporte une contribution importante. Il devient possible, à partir de la structure d’une protéine, de sélectionner les petites molécules pouvant interagir avec elle. L’étude in silico évite de coûteuses expériences in vitro. Le calcul de certaines propriétés des molécules (taille, masse, nombre de liaisons hydrogènes…) permet d’indiquer celles qui pourraient être absorbées par voie orale. On peut aussi cribler dans l’ordinateur un grand nombre de candidats, afin de sélectionner ceux qui ont le plus de chance d’interagir avec une cible thérapeutique particulière.
Au cours des trente dernières années, notre compréhension de la structure des molécules a beaucoup progressé. Dans un futur proche, on pourra sans doute aborder, au travers de nouveaux outils de perception (visualisation, robotique, impression 3D, etc.), des problèmes d’une complexité supérieure, comme la simulation de la diffusion des molécules et la formation des assemblages moléculaires impliqués dans les cellules entières. Avec la perspective de nouvelles retombées pharmaceutiques et biotechnologiques.
Livres
- Branden C., Tooze J., Introduction à la structure des protéines, De Boeck, 1996.
- Fourme R., « Imager les molécules du vivant », in DocSciences, n°4, CRDP de Versailles, 2008.
Sites web
Cet article est paru dans la revue DocSciences n°8 Le numérique et les sciences du vivant, éditée par le CRDP de l’Académie de Versailles en partenariat avec l’Inria/a>.
Niveau de lecture
Aidez-nous à évaluer le niveau de lecture de ce document.
Votre choix a été pris en compte. Merci d'avoir estimé le niveau de ce document !

Pierre Tuffery